Introduction
Cystic fibrosis (CF) affects mainly the lungs and pancreas; however, there is also a risk that organs of the digestive and reproductive systems, as well as kidney and bladder, will be damaged (Di Lullo et al. 36). With the development of the disease, the patient acquires different symptoms, and his/her state becomes worse. The typical signs of CF might include coughing, increased mucus production, diarrhea, diabetes, weight loss, and salty sweat (Bisch et al.). The emergence of symptoms and their severity depend on the organ affected by CF and the stage of the disease.
Main body
At the cellular level, CF causes thick mucus to build up and deteriorate the functioning of different parts of the body, such as the lungs (Jacob et al.). The major cause of the emergence of this problem and the buildup is the gene called cystic fibrosis transmembrane regulator (CFTR), which controls the flow of water and salt in and out the cells of the body (Standen 40). The emergence and development of CF mean that mucus becomes thickened, which influences the work of cells and parts of the body.
CF genes
From the given pedigree, it is possible to see that both Sarah and Michael’s parents were carriers of the CF gene. However, their children acquired different patterns. Sarah’s sister became affected by CF and died, while Sarah did not get the disease allele. The same can be stated about Michael. His brother became sick with CF and deceased, while Michael avoided acquiring CF allele.
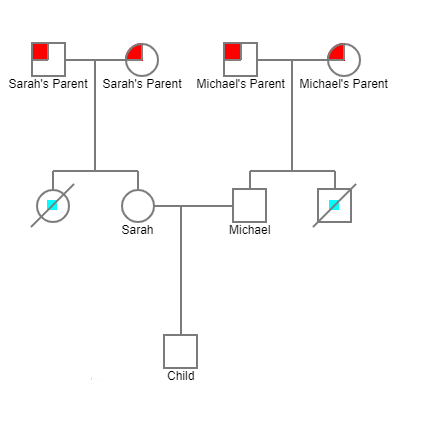
From the pedigree, it is possible to conclude that Michael and Sarah were not affected by the disease, although their parents were all carriers of the CF allele (Freeman et al. 317). Instead, their sister and brother acquired CF because of the existence of recessive genes in their parents. In such a way, the gender factor is not relevant for the case as Michael and Sarah avoided being affected. The autosomal recessive inheritance can be observed here. However, if gender impacts the transmission of genes, x-linked dominant or x-lined recessive types can be stated (Tluczek et al. 289).
Punnet Scale
The following Punnet scale can be offered, assuming that Sarah is not a carrier. The table is created using information about parents. Male genes are displayed at the top, while female ones are on the left (Freeman et al. 305). In such a way, there is a 50% that a child will be a carrier of CF and 50% that a child will have the disease. It comes from the table as there is an equal distribution of chances.
If Sara is considered a carrier, the situation becomes different. Using the Punnet scale, it is possible to predict that she and her husband can have a baby with CF 1 in 4 cases. In such a way, it becomes critically important to determine the presence of the gene in both parents to predict the risks and the opportunity to have a healthy child.
Results of the Analysis
Both Sarah and Michael have their siblings lost, which means that their parents must be carriers. Moreover, it means that there is a 2 in 4 chance that both a male and a female are carriers and might have a child with CF, depending on the combination of genes. In such a way, the situation will not change much as both individuals have a similar position and are both vulnerable to the disease. As far as there is no dependency on gender in this sort of inheritance, Michael and Sarah have the same chances of being carriers or having children with the given disease.
References
Bisch, Alexander, et al. “Cystic Fibrosis Transmembrane Conductance Regulator Genotype, Not Circulating Catecholamines, Influences Cardiovascular Function in Patients with Cystic Fibrosis.” Clinical Medicine Insights: Circulatory, Respiratory and Pulmonary Medicine, 2019, Web.
Di Lullo, Antonella, et al. “Cystic Fibrosis: The Sense of Smell.” American Journal of Rhinology & Allergy, vol. 34, no. 1. 2020, pp. 35–42, Web.
Freeman, Scott, et al. Biological Science, Third Canadian Edition. Pearson Canada. 2018.
Jacob, Anne, et al. “How Do Cystic Fibrosis Patients Experience Parenthood? A Systematic Review.” Journal of Health Psychology, 2020, Web.
Standen, Jen. “Cystic Fibrosis.” InnovAiT, vol. 13, no. 1, Jan. 2020, pp. 39–46, Web.
Tluczek, Audrey, et al. “Impact of Intermediate Cystic Fibrosis Classification on Parents’ Perceptions of Child Vulnerability and Protectiveness.” Journal of Family Nursing, vol. 25, no. 2, 2019, pp. 287–313, Web.