Gene mutations are changes in the nitrogenous base pairs resulting in the false expression of protein due to false translation and transcription processes. Genetic mutations could be due to substitution, addition, deletion, inversion of base pairs that are caused by chemical or environmental factors. Genotypic alterations result in a change in the expression of proteins, thus, altering the phenotypic response.
One of the most typical and lethal demonstrations of a genetic mutation observed in the current era is that of ‘Progeria’- a disease that causes premature aging in infants (Secerbegovic, 1997). This disease is found once in every 4 million births and caused death usually by the early teens. This disease is caused by the mutation of a lamin-A gene, (LMNA); single nucleotide mutation is observed in this gene, thus causing autosomal–dominant expression in patients, which means that it is a ‘chance’ mutation, and will not be passed down in families.
As depicted in the studies conducted by Kris and his co-workers (2006), this segmental premature aging disease is caused by Hutchinson-Gilford Progeria syndrome (HGPS), named after the two scientists who discovered it in 1886. Travis (2003) describes that “Children with progeria are usually diagnosed 6 months to a year after birth when their physical development starts to lag. They rarely grow taller than 4 feet, and their heads are oversized for their bodies. The children become bald and have skin problems such as scleroderma. While their mental development is normal, children with progeria rapidly develop atherosclerosis and die, on average, at the age of 13”. Balding of the head and narrowing of eyebrows are observed, along with sunken eyes, a bead-like nose, and prominent head veins. They are observed to die of the same causes as observed in the old, like, cardiac arrest, high blood pressure, and are prone to have dislocated or loose hip joints, with atherosclerosis, as observed by Mazereew (2007).
The discovery of the gene that causes this fatal disease was discovered only in the recent past when a group of scientists working in the Progeria research foundation in the year 2003 as cited by many scientists and published in Nature, science, and many prestigious journals, (Mazereew, 2007). This promising revelation revolutionized and excited the scientific community, as a ray of hope was seen in curing or reversing this mutation, once the gene was identified.
As reported by Shurong (2005), Progeria is caused by a de novo single point mutation, by which the structural and mechanical properties of the nuclear lamina are altered. Specifically, a mutation in the Lamin A and C regions is observed due to the single point deletion of the nucleotide array. In his study, Shurong describes that “Lamin A protein is observed to be the structural scaffolding of the nucleus that holds it together, and is also involved in gene expression and DNA replication”.
A formidable study was conducted by Mark and his coworkers (2007) demonstrating the new approaches to Progeria, in which he shows by experimentation that out of the 664 amino acids being translated onto mRNA, the prelamin gene at exon 11 undergoes single nucleotide substitution, such that GGC becomes GGT at position 1824. He further explains in the paper that this mutation does not alter the protein sequence, as it is a ‘silent’ mutation; however, it results in the deletion of a 150- nucleotide stretch of the exon. This missing chain sequence causes the lethal disease by shrinking the laminar membrane of the nuclei.
These abnormalities can be ‘corrected’ employing RNA interference, as shown by Shurong. The ‘missing’ 150 base pair region can be manipulated in order to provide sufficient tensile strength to the nuclear membrane. However, lots of research inputs are required to further explore these options. Likewise, scientists are now studying the genetic mutations in LMNA of centenarians and comparing these mutations with patients who have progeria (http//www.progeria-research.org).
There is a dire need to dig deep in deciphering the exact cause as to why this mutation takes place and how it could be rectified. Until and unless resources are poured in, the tiring efforts by these talented researchers who have come so far will go to waste, and millions of children will continue suffering in silence. Even in this modern era, where science and technology have made quantum leaps toward its progress, it is indeed very heartbreaking to see the innocent die due to our ignorance. The scientific community should prioritize its objectives, in first saving young innocent live, enormous resources and efforts should be galvanized in solving this problem, as to how the disease could be ‘reversed’, this will definitely be a breakthrough advancement, where science would have actually saved lives. World wide programs should be launched, and with the huge potential available, in terms of scientific talent, efforts should be pooled, and it is high time that this lethal disease that has been prevailing for so long, killing millions each year, should be looked into, and be placed as number one, on every research agenda.
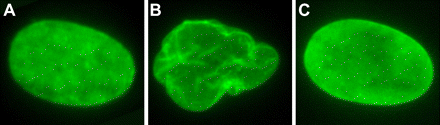
“Cultured human fibroblast nuclei demonstrating nuclear morphology. An Untreated normal control fibroblast. B, Untreated progeria fibroblast. C, FTI-treated progeria fibroblast 36 hours after treatment” (Kieran et al, 2007).
References
- S. Secerbegovic (1997) A hypothesis that aging results from defects in genetically produced proteins Medical Hypotheses 48, 531-533
- Shurong Huang, Lishan Chen, Nataliya Libina Joel, Janes, George M. Martin, Judith Campisi, Junko Oshima (2005) Correction of cellular phenotypes of Hutchinson-Gilford Progeria cells by RNA interference Hum Genet (2005) 118: 444–450
- Mark W. Kieran, Leslie Gordon, and Monica Kleinman, (2007) New Approaches to Progeria-PEDIATRICS Vol. 120 No. 4 2007, pp. 834-841.
- Progeria Research.
- Pediatrics.
- Kris Noel Dahl Paola Scaffidi, Mohammad F. Islam, Arjun G. Yodh, Katherine L. Wilson, and Tom Miste (2006) Distinct structural and mechanical properties of the nuclear lamina in Hutchinson–Gilford progeria syndrome PNAS vol. 103 no. 27,10271-10276
- J. Mazereeuw-Hautier, L.C. Wilson, S. Mohammed, D. Smallwood, S. Shackleton, D.J. Atherton and J.I. (2007) Harper Hutchinson–Gilford progeria syndrome: clinical findings in three patients carrying the G608G mutation in LMNA and review of the literature PAEDIATRIC DERMATOLOGY j.1365-2133.2007.07897.