Iron regulation in the body
Iron is a constituent of all living matter. Iron is a core factor in the electron transfer chain in the body; it is also a vital part of oxygen transport and iron storing molecules such as hemoglobin. It is also a component of host defense where it occurs in nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. Humans lack an effective mechanism to excrete surplus iron, though they are unsurpassed in iron conservation since they have unique homeostatic mechanisms that ensure iron level remains within normal parameters. These mechanisms regulate the absorption of iron from the duodenum and release it from macrophages and other iron-storing sites. The release of large amounts of iron into circulation may result in localized injury to the surrounding tissues. Iron exerts its toxic effects through the catalysis of free radical reactions leading to the production of free radicals such as peroxides that damage tissues. Furthermore, iron levels need to be maintained within physiological limits since infection is in part determined by the availability of iron for utilization by the invading microorganism. The bacteria have evolved complex mechanisms for uptake of iron from their environment that bind iron. An example of such mechanisms includes the secretion of organic molecules called siderophores that have a high affinity for iron. Desferrioxamine an iron binder is an example of such a siderophore. Other siderophores include yersiniabactin produced by Yersinia sp (Collins 2003, p.194).
Studies carried out show that mice with iron overload are highly susceptible to a wide range of infections. This phenomenon has also been observed in human patients that have iron overload. (Jurado 1997, p. 888-890). Since little iron is lost from the body, iron regulation is through stepwise regulation of its absorption from the gut into the general circulation. Iron in the body is usually recycled and stored in various body stores (Ganz & Hershko 2000, p.2-4). Amplification of the absorption process occurs in states of iron deficiency whereas absorption is decreased in situations of iron overload. Through this mechanism, the body is able to maintain iron levels within normal limits (Finch et al 1978, p.335). Regulation of iron is an essential process since an excess of iron leads to end-organ damage while deficiency of iron results in cellular dysfunction and anemia. Regulation of the body’s iron stores is under the influence of specific iron regulating proteins. These proteins include transferrin, ferritin, ferroportin, and Hepcidin. Other proteins such as lactoferrin control iron levels through chelation to form iron-protein complexes (Waheed et al 1999, p.15).
Total body iron
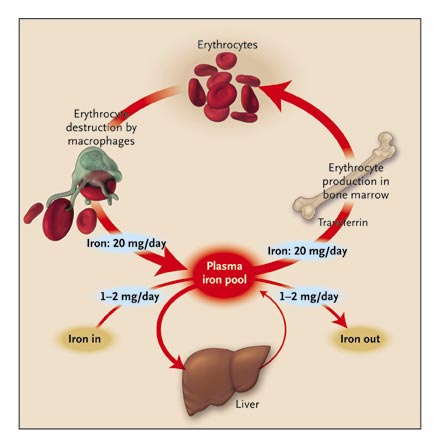
A normal human body contains about 4 to 5g of Iron. Haemoglobin iron forms the bulk of total body iron, accounting for up to 60% of total body iron. Hemoglobin is major the functional form of iron in the body (Harris & Kellermeyer 1970,p 87). Approximately 10% of total body iron exists in the body as ferritin and hemosiderin. The liver stores iron as ferritin and this accounts for a third of total body iron. The remainder of iron is stored throughout the body tissues as myoglobin and in heme enzymes such as catalases, cytochromes, and peroxidizes and non-heme enzymes such as reductase, metalloflavoproteins, and ribonucleotide reductase. The stored iron exists in macrophages that populate the spleen, liver, and bone marrow. Iron occurs in minute quantities in circulation usually about 3-4mg (Donovan et al 2000, p.776-779). When red blood cells become senescent, they undergo lyses and liberate iron, which is incorporated in the synthesis of hemoglobin and other iron-dependent proteins. About 20mg of iron is recovered through this process. Daily losses of iron from the body are in minute quantities. It is estimated to be around 1-2mg of iron, which is readily replaced from the diet (Bothwell 1995, p.24)
Iron Absorption
The brush border of the epithelium of the intestinal villi of the duodenum and upper jejunum is the site of iron absorption in the human body. Iron is absorbed in form of heme, non-heme, ferric, or ferrous ions. Studies show that at least nine proteins are involved in the absorption process (Bothwell 1995, p.28). The amount of iron available in the body has an inverse relationship with the amount of iron absorbed. When body iron stores decrease, the amount of iron absorbed increases. When there is iron overload, the absorption of iron decreases. Dietary iron is mainly non-heme, which is readily absorbed depending on the presence of inhibitors or promoters. Inhibitors such as phytates and tannins decrease iron absorption while promoters such as ascorbic acid enhance absorption.
Mucosal cells regulate iron absorption through various mechanisms: regulation of uptake across the brush border, retention of iron in the mucosal cell in a different form, and release of iron from the mucosal cell into plasma (Avunduk 2008, p. 427). Meat-derived iron is absorbed four times faster than vegetable-derived iron. An average adult consumes approximately 10 to 15mg of iron daily. Women lose more iron than men do. Iron is lost as “exfoliated epithelial cells of the gastrointestinal tract and skin, and menstrual blood loss in women” (Avunduk 2008, p. 427). The body lacks any physiological mechanism to excrete iron. As such, iron absorption should be finely controlled and regulated within the narrow physiological range.
Iron Control Centre
The liver is the center of control of iron homeostasis in the body. Control of iron levels in the body is essential since an overload results in organ damage and conditions like hemochromatosis. The deficiency of iron results in anemia usually referred to as iron deficiency anemia. The iron regulation process employs specialized sensors. The author writes,
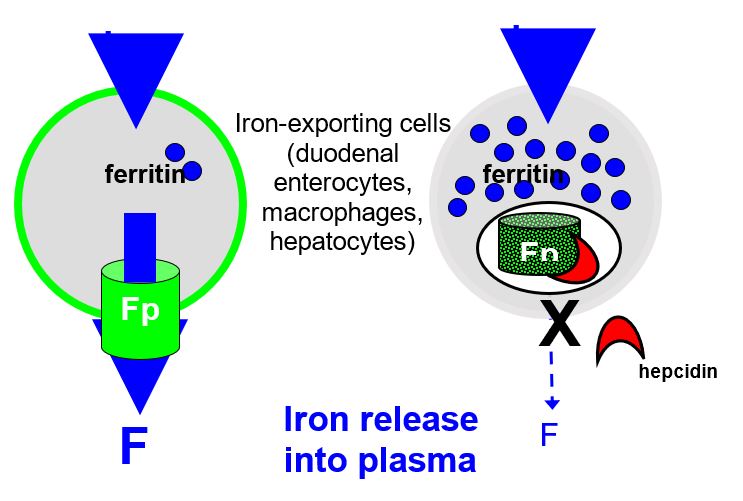
“Special sensors respond to the iron and stimulate the synthesis and release of the iron hormone Hepcidin, which is encoded by HAMP gene. Hepcidin circulates through the body and interacts with the iron exporter ferroportin expressed on the surface of iron-rich macrophages and intestinal cells. Because of this interaction, ferroportin is internalized and degraded. The unneeded iron remains in the cells where it is saved for future use in the form of ferritin. The diminished release of iron restores blood levels to the non-toxic range, thus reinsuring the stimulus for further Hepcidin synthesis and ferroportin gradually resumes its iron exporting activity”( Avunduk 2008, p. 428).
The connection between Hepcidin and iron overload was first brought to light by Pigeon et al during the study of the response of the liver to iron overload. (Pigeon, Ilyin & Courselaud 2001,p. 16). They found out that the Hepcidin mRNA was induced by an iron overload of parenteral and dietary origin. Furthermore, they found out that induction of Hepcidin mRNA occurred when mice were treated with polysaccharides (LPS) to simulate an active bacterial infection. From these results, it was concluded that Hepcidin production was under the influence of iron and an immune system stimulus (polysaccharide). It was also observed that the Hepcidin gene is in the same loci as the upstream regulatory factor-2(USF-2) gene. Other gene knock-out studies reinforced the relation between USF-2gene and iron overload since mice that had their USF-2 gene knocked all developed hemochromatosis at the same time. (Nicholas, Bennoun &Devaux 2001, p. 12-14). It was further observed that these mice had no Hepcidin mRNA expressed. This led the authors to conclude that Hepcidin was a controller of iron absorption from the duodenum and upper jejunum and that it was a regulator of iron release from macrophages. It is the absence of Hepcidin that resulted in iron overload (hemochromatosis) since the gene coding for the Hepcidin gene is located in the same loci as the USF-2 gene.
Mechanism of action of Hepcidin at the molecular level
Inflammation and iron overload induce transcription of Hepcidin mRNA in the hepatocytes. Inflammatory mediators induce the production of cytokines such as IL-1, IL-6, TNF-β, and TNF-α. IL-6 acts by amplifying the transcription process. Hepcidin is produced in high amounts and released into circulation to be delivered to iron exporting cells located in the small intestine and spleen. Hepcidin then binds to the iron exporting protein called ferroportin (Fpn). When Hepcidin binds to ferroportin, it leads to activation of the Jak2 pathway and subsequent phosphorylation of ferroportin. The phosphorylated receptor is turn internalized and degraded by the lysosome hence impairing iron export from the affected cell into circulation (De Domenico 2007, p. 2569-2578.) It has been established that in absence of iron overload and inflammation, macrophages have little ferroportin on their surface.
Importance of Hepcidin
Hepcidin is an essential component of the body’s homeostasis process. It is the main regulator of iron homeostasis in the body. Furthermore, it plays a role in the innate immune system. Many iron disorders are a result of abnormalities in Hepcidin production. Production of little amounts of Hepcidin causes iron overload in conditions like hereditary hemochromatosis and β-thalassemia a form of anemia due to a defect in one of the globin chains. On the other hand, excessive production results in anemia of inflammation initially called anemia of chronic disease (ACD). In anemia of inflammation, there is pronounced sequestration of iron in the macrophages while in thalassemia there is excessive breakdown of erythrocytes causing a release of massive amounts of iron that may result in overload even in the absence of blood transfusions.
Discovery of Hepcidin
Hepcidin was discovered when Park et al were conducting studies on antimicrobial characteristics of various fluids in the human body (Park, Valore & Ganz 2001, p. 7806-7810). They extracted a new peptide from urine and named it Hepcidin based on its site of origin (hep- from hepatic) and its antimicrobial activity (-cidin; to kill). Krause et al working separately extracted a peptide with similar properties and referred to it as LEAP-1 (liver expressed antimicrobial peptide) which later on was found to be Hepcidin. (Krause, Neitz & Magert 2000,p.147-150).From these studies, it was learned that Hepcidin is composed of 25 amino acid residues and 4 disulfide bridges connecting the residues as shown in the diagram.
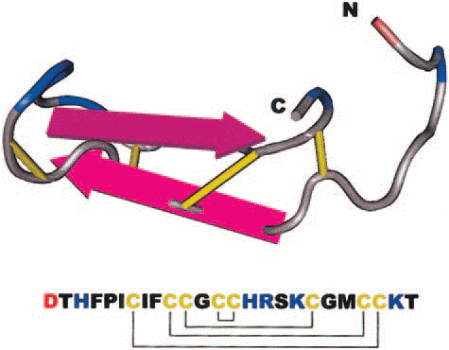
The amino terminal corresponds to N while carboxyl terminal corresponds to C.Disulfide bonds, acidic amino acids and basic amino acids are represented by the colours yellow, red and blue respectively.
It was noticed that in many mammalian species the Hepcidin peptide sequence was astonishgly analogous. Hepcidin arises from an 84- amino acid prepropetide, which is encoded by a gene on chromosome 9. Drosomycin, a defensin of drosophila insect, released during infection is almost analogous to Hepcidin. It is composed of 4- disulfide bond within its structure. These cysteine rich residues are responsible for the defensive antimicrobial properties of drosomycin. Drosomycin production occurs in the fatty layer of the insect an equivalent of the liver in mammals. Studies have also shown an increase in Hepcidin levels in patients with active infection (Gangaidzo, Moyo & Mvundura 2001, p. 936-939).
Production of Hepcidin
Hepcidin is produced predominantly by hepatocytes. The Hepcidin mediated activities; iron regulation and antimicrobial activity are under the influence of one gene unlike in the mouse where we have two Hepcidin genes but only one code for iron regulation (Pigeon 2001, p.7811-7819)
Regulation of production
Effect of Iron on production Hepcidin
Homeostatic regulation of Hepcidin production is under the influence of iron levels and the state of erythropoiesis. Hepcidin production is upregulated when there is iron overload and downregulated in situations of iron deficiency. At the same time pronounced erythropoiesis results in decreased production of Hepcidin to allow for increased absorption and release of iron from macrophages to meet the demands of the body.
Production is also high in inflammatory conditions and infections. Although precise mechanisms of regulation of production of Hepcidin have not been elucidated, it is widely believed that regulation is under the influence of factors released by the bone marrow. Such factors include the bone morphogenetic protein (BMP). This protein has been demonstrated to stimulate increased production of Hepcidin both in vitro and in in vivo (Xia, Babitt, & Sidis 2008, p. 5195-5200). Several BMPs have been identified but BMP-6 has been repeatedly demonstrated to be the dominant regulator of Hepcidin production (Andriopoulos, Corradini & Xia 2009, p. 482–487). Activation of BMP-6 occurs via co-receptors.
Hemojuvelin (HJV) has been identified to be the co-receptor that modulates BMP-6 activity. Mice that have HJV mutations develop iron overload without any other noticeable abnormality. Studies carried out in recent times have shown that in mice with iron overload there is an increased expression of the BMP-6 mRNA linking BMP-6 to be an indicator of the amount of iron in the body. Some scientists have proposed that interfering with the BMP-6 pathway may be of therapeutic value in cases of Hepcidin excess.
The transferrin receptor 2(TfR2) and hemochromatosis gene (HFE) are the iron level sensing genes and in hereditary hemochromatosis, these two are mutated. TfR2 is of hepatic origin, the source of Hepcidin. The iron-transferrin complex binds to TfR2 and stabilizes it. The new complex causes increased signal transduction of ERK1/2 and smad1/5/8(Goswami & Andrews 2006, p.28494–28498). HFE and iron transferrin share a site of association on TfR1 and when the levels of iron-transferrin complex are high, HFE is displaced from the site of association on TfR1. Despite the above, studies have repeatedly demonstrated that HFE and TfR2 play no role in iron regulation mediated by Hepcidin(Piperno, Girelli & Nemeth 2007,p.24-25).
Effect of Erythropoiesis on production
Elevated erythropoiesis is a major inhibitor of Hepcidin production. A study carried out in mice showed a dose-dependent decrease in expression of Hepcidin mRNA hours after they received a specified dose of erythropoietin. However, the effect of erythropoietin on the production of Hepcidin is not well defined (Ashby, Gale & Busbridge 2007, p.18). The mechanism by which erythropoiesis inhibits Hepcidin production is not clearly understood. It has been postulated that proteins related to TGF-β superfamily are involved in this process. These proteins include growth differentiation factor 15 (GDF15) and twisted gastrulation protein (TWSG1). TWSG1 binds BMP-6 leading to suppression of expression of Hepcidin. This reinforces the theory that Hepcidin regulation is mediated via the BMP-6 pathway.
These proteins were also detected in high levels in dyserythropoietic anaemias and β-thalassemias. The mechanism by which hypoxia suppresses Hepcidin production still remains to be determined.
Effect of inflammation on Hepcidin production
Inflammation is a potent stimulator of Hepcidin production. Studies have shown a rapid increase in levels of Hepcidin within the onset of infection and inflammation in both mice and humans. Hepcidin decreases the export of iron from macrophages hence depriving the microbes of iron, an essential component for the survival of the microbe (Sharma, Nemeth & Chen 2008, p.3262-3267). Inflammatory cytokines, specifically IL-6 mediate the up-regulation in Hepcidin production (Nemeth et al 2003, p. 2461-2463). In inflammation hepcidin production is greatly increased. !00-fold increase of hepcidin was recorded during inflammation due to more virulent organisms. Small increases in production were observed when inflammation was due to less virulent microorganisms. Patients with sickle cell anaemia and myelodysplasia had a similar magnitude of hepcidin increase after they were transfused with fresh blood. Studies carried out n isolated human hepatocytes revealed an increase in hepcidin mRNA production when the hepatocytes were exposed to lipolysaccharide. This was evidence that hepcidin production was under influence of inflammation. This is explained as follows: the lipopolysaccharide molecules interact with macrophages, in this case, hepatic kuppfer cells. The kuppfer cells are activated to release cytokine messengers such as Interleukin-6(IL-6), a mediator of inflammatory response. IL-6 then induces hepcidin mRNA transcription and subsequent hepcidin production. From the above, it is clear that hepcidin plays a major role in inflammation.
Hepcidin and haemochromatosis
Haemochromatosis is a cluster of inherited conditions that have a common feature of excessive iron absorption resulting in iron overload. Mutations in the genes that regulate various aspects of iron metabolism are responsible for this condition. Mutations in the haemochromatosis gene (HFE) are linked to most cases of haemochromatosis especially in people of Caucasian descent (Acton, Barton & Snively 2006, p. 815-821).
Advances in genetic studies link Hepcidin to most forms of hemochromatosis in humans. Examples include HFE-hemochromatosis, Transferrin receptor 2, Juvenile hemochromatosis (HJV), Juvenile hemochromatosis (HAMP), and Ferroportin disease (SLC40A1). In haemochromatosis, Hepcidin production is impaired or in some cases, there is Hepcidin resistance. As a result, expression of ferroportin on the basolateral membrane is amplified and subsequent increase in rate of absorption of iron across the enteric wall occurs leading to iron overload. At the same time, absence of Hepcidin results in increased export of iron from macrophages into general circulation (Fillet, Beguin & Baldelli 1989, p. 844-851).The high levels in circulation lead to end organ damage and may result in death if appropriate medical intervention is not initiated in time.
Animal models with defective Hepcidin production mechanism develop haemochromatosis. This phenomenon is attributed to a mutation of the human haemochromatosis protein (HFE) gene, a component of the major histocompatibility complex (MHC) class 1. Studies have shown that mice that have their HFE gene knocked develop iron overload but the expected increased transcription of Hepcidin mRNA does not occur (Bridle, Frazer &Wilkins 2003, p. 669-673). Corradini, Garuti & Mothosi (2009, p.1489-1490) brings to light the link between BMP-6 and HFE. Mice with impaired HFE gene expression exhibited a dysfunctional BMP-6 signalling pathway).
Hepcidin as an antimicrobial peptide
Hepcidin is composed of 25 amino acid residues with 4-disulfide bonds within its structure. It is a derivative of an 84-amino acid prepropetide produced in the liver. Other smaller Hepcidins with antimicrobial properties have also been isolated from human urine. These are the 20 and 22- amino acid residue Hepcidins. These Hepcidins have also been shown to have antimicrobial activity against a wide range of microbes. Hepcidin was named so in regard of its antimicrobial properties i.e. –cidin meaning to kill and hep-, in regard to its site of origin.
Studies have shown that there is an increase in transcription and translation of Hepcidin mRNA during infection and inflammation.. This led scientists to the theory that Hepcidin is an antimicrobial agent. Analogues of Hepcidin in other species have reinforced this phenomenon. An example is drosomycin, which is produced by the insect drosophila in its fatty layer (an equivalent of the liver in humans) during acute infections. Drosomycin acts as a defensin against bacterial infections in these insects. Other insect defensins exhibiting Hepcidin like activity include heliomicin and thanatin (Fehlbaum, Bulet & Michaut 1994, p. 33159–33163). Hepcidin is the equivalent of these insect defensins in humans.
In studies carried out Hepcidin demonstrated cidal activity against a wide range of microbes. Examples include Candida albicans, Aspergillus fumigatus, Aspergillus niger and antibacterial activity against both gram positives such as Staphylococcus aureus, Staphylococcus epidermidis, group B Streptococcus and gram negatives bacteria such as Escherichia coli( Akarsu & Mamuk 2007,p.1849-1850)
Hepcidin mRNA transcription is induced by liposaccharide(LPS) both in vivo and in vitro which is produced by invading microbes in the natural environment (Yang, Chertov & Bykovskaia 1999, p.525–528). Scientists have postulated that Hepcidin acts through binding on the cell wall surface of microbes. The antimicrobial activity is mediated by absence of iron for utilization by the invading organisms. When a microbe invades the host it initiates production of IL-6 which in turn amplifies transcription of Hepcidin mRNA. This leads to release of Hepcidin from hepatocytes into circulation. Circulating Hepcidin finds its way to the splenic macrophages, enterocytes and other iron containing cells where it binds to ferroportin an iron exporter inducing its internalization and subsequent degradation. As a result of this, iron export from these cells is impaired hence depriving the invading microbes off an essential component required for their survival.
It is further proposed that Hepcidin acts by penetrating invading cells through toll-like receptors and lead to accumulation of iron inside the microbe. When accumulated iron exceeds the level required by the microbe it becomes toxic to the microbe resulting in organ damage and death of microbes. Similarly transferring an important iron transporter is closely related to lactoferrin, a potent iron chelator found on the surface of neutrophils and in secretions of the epithelium. Antimicrobial activity of lactoferrin is in part attributed to its iron chelation properties.
Divalent metal trasporter 1(DMT1) is the main mechanism through which iron is absorbed across the intestinal wall. This transporter is analogous to Nramp 1, a cationic transporter found on the walls of macrophages. Mutations in the gene coding for this transporter eliminates the ability of macrophages to attack and destroy pathogens leading to an increased tendency of having infections. (Forbes & Gros 2001,p.397-403).
Anaemia of Inflammation
Anaemia of inflammation initially referred to as anaemia of chronic diseases ( ACD) is a normocytic normochromic anaemia that occurs in individuals who have an infection, neoplasm or inflammatory condition for a period of 1 to 2 months. It is characterized by low iron levels available for utilization in presence of sufficient iron in various storage sites within the body. (Lee 1983, p. 10).Anaemia of inflammation is associated with several conditions that are infectious, non-infectious or malignant in nature. Anaemia becomes evident within the first 2 months of onset of the chronic illness. It occurs when we have infections due to bacterial microbes,viral and even yeasts. Anaemia of inflammation also occurs in autoimmune disorders such as rheumatoid arthritis and systemic lupus erythematosus. Chronic diseases that have a slower course of progression such as, chronic kidney disease (CKD), liver failure, cancer and heart failure have been found to cause anaemia of inflammation as well as pro-inflammatory states such as aging.
Though anaemia of inflammation is usually identified as normocytic normochromic anaemia, it may progress to become microcytic hypochromic type of anaemia as the disease advances. The phenomenon of anaemia of inflammation may be a protective mechanism by the body against infections from iron dependent microbes. In anaemia of inflammation, macrophages are devoid of iron, transferring saturation is at its lowest and there is low levels of iron in serum.
In this type of anaemia, the bone marrow’s response to erythropoietin (Epo) is diminished. This results in reduction of the lifespan of available red blood cells and increase in rate of breakdown of senescent ones in the spleen. This is a paradoxical occurrence since in anaemia of inflammation, the body aims to protect itself from the harmful effects of excess iron however, at the same time, the sequestration of iron results in shortened lifespan of the red blood cells.
Pathogenesis
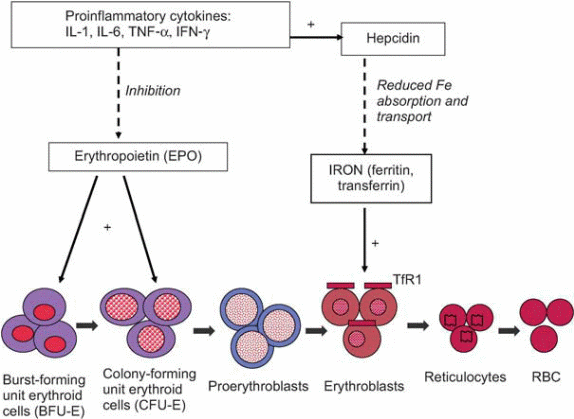
Most of these conditions are associated with the production of cytokines such as IL-1, IL-6, and TNF α and β. In this type of anemia, iron levels in the blood are decreased within 24 hours of the onset of active infection. Its been postulated that the low blood levels of iron can be attributed to a shift of transferring-bound iron to the ferritin form which is not available for utilization by the body( Dallalio, Fleury & Means 2003,p.4). IL-1 amplifies ferritin mRNA translation leading to the production of large quantities of ferritin that bind iron. Cytokines such as IL-6 stimulate the production of Hepcidin, an acute-phase protein produced by the liver. Hepcidin is believed to decrease absorption of iron from the small intestine and release of iron from macrophages resulting in low iron levels observed in anemia of inflammation. Studies show a strong correlation between levels of Hepcidin in urine and blood level of ferritin. Patients with anemia of inflammation have high levels of Hepcidin in their urine in comparison with patients who have iron deficiency conditions (Means & Krantz 1992, p.1639-1647.) Other cytokines such as TNF-α inhibit the expression of Hepcidin in mice and humans resulting in iron overload and hemochromatosis.
Hepcidin associates with its receptor, ferroportin on macrophages and enterocytes preventing them from releasing iron into circulation. When Hepcidin binds ferroportin, it initiates phosphorylation of ferroportin and subsequent uptake into lysosomes (internalization) where it undergoes degradation. This process is mediated via the JAK2 signal transduction pathway. As a result, the iron exporter is not expressed on the surface of macrophages and duodenal enterocytes. Due to this, the export of iron from these cells into general circulation is impaired and iron remains within these cells leading to hypoferremia. Ferroportin is the only transporter associated with the export of iron from cells as a result it provides an essential target for agents that can be used in the management of the anemia of inflammation (Weinstein & Roy 2002, p. 3776-3781).
Studies have also demonstrated a unique feature of erythrocytes in patients with anemia of inflammation. The erythrocytes in such patients have a shortened lifespan with a high turnover, though the iron released from their breakdown is sequestered by macrophages leading to a hypodermic state though iron is available within the body (Gabrio, Finch & Huennekens 1956, p. 103-104)
In chronic conditions, there is a large pool of cytokines in circulation and these cytokines induce proliferation of macrophages which in turn cause phagocytosis of the erythrocytes leading to anemia of inflammation. This action is mediated through cytokines like TNF-α, an inhibitor of Hepcidin transcription (Card 1988, p.40-43)
Other pathogenetic factors implicated in anemia of inflammation involve a wide range of interleukins that are inhibitory to erythroid series proliferation. Of this interferon-γ exerts more inhibitory influence on the series. The inhibitory effect brings about a downregulation of Epo associated receptors and apoptotic effect on the erythrocytes.
Effect of Hepcidin on the innate immune system
The innate immune system is a primordial defense mechanism that exists in mammals and other species including those in the plant kingdom. This system has two very important functions that are essential for defending the host against invading pathogens. First, it offers a physical barrier to invading microorganisms and secondly it primes the adaptive system with instructions on how to handle the invading microorganism.
Scientists have termed this system crude since it offers a “blanket cover” to the body from any invading microorganism regardless of its nature. The innate immune system is composed of various elements that afford it the “blanket cover” when acting against invading pathogens. It employs a myriad of cells to defend the body. These include natural killer cells, macrophages (which interact with Hepcidin), dendritic cell precursors, neutrophils, basophils, tissue mast cells, and epithelial cells.
This system has evolved in order to differentiate between normal components that occur in the environment and pathogens. To achieve this, vertebrates encode special molecules called PAMPS. PAMPS are vital molecular structures that occur in almost all pathogens and are essential for the pathogen to maintain and exert its virulence when attacking the host immune system.
The vertebrates have further evolved to generate molecular components to augment the innate or nonspecific immune system. An example of such molecules is Hepcidin, a 25 amino acid peptide produced by the liver in mammals. Hepcidin is an iron-regulating hormone that is released during infection or in presence of high levels of iron in circulation. The release of Hepcidin in such circumstances has been a core point of study by scientists (Douglas, Gallant & Liebscher 2003, p.589–601.)
Studies show that microorganisms require iron in order to survive and exert their virulence. These microorganisms have evolved complex mechanisms to extract iron from their environment. They produce complex molecular compounds called siderophores that bind any available iron for their utilization (Winkelmann 2002, p.691–696.) These molecules also extract iron from their storage sites such as ferritin and lactoferrin. The bound iron is transported into the bacterial cell via various transport mechanisms such as the protein-dependent transport complexes, PBTs (Faraldo-Gomez & Sansom 2003, p. 105–116.)
It has been demonstrated that the PBTs act by leading the siderophore-associated iron to the ferric reductases on walls of the microbe. Furthermore, the PBTs internalize the siderophore-associated iron making it available for utilization by the pathogen. In the presence of Hepcidin, all these iron acquiring mechanisms by the microbe are inhibited and as such, Hepcidin has been appreciated to be a main component of the innate immune system in mammals.
Further studies have shown that those microbes that fail to express siderophores for iron extraction have reduced virulence. An example is the V. Vulnificus bacteria that do not express the catechol-derived siderophore. Consequently, it has reduced bacterial virulence as compared to the other bacteria in the same genus that express this type of siderophore due to lack of iron (Weinberg 2000, p.85-89). Furthermore, V. Vulnificus has a mutation in one of its metalloprotein components that hinders it from obtaining iron it’s transporting protein transferrin and lactoferrin. These observations have led researchers to be convinced that iron plays a crucial role in the virulence of V. Vulnificus and other microbes as well. Microbes such as Yersinia species rapidly proliferate and reproduce in body fluids that are rich in iron and macrophages, which are rich in iron. The most virulent strains of Yersinia species have a gene that encodes for a potent iron siderophore called yersiniabactin (Carniel 2001, p.561-569.). This behavior occurs in other enterobacteria such as E. coli, Klebsiella sp, Citrobacter sp, and Salmonella enterica.
Iron is an essential component for the survival, growth, and infectivity of a microbe or pathogen. Hepcidin is the main iron regulating peptide in the body and in its there is the availability of iron to microbes hence leading to their unhindered proliferation. However, Hepcidin upregulation through a series of cytokine-mediated processes facilitates decreased absorption of iron and further sequestration in macrophages hence making it unavailable for utilization by the pathogens. Studies show that mice with a mutation in the genes coding for Hepcidin result in increased susceptibility of these mice to infections. The infections are widespread ranging from both gram-positive and gram-negative bacteria to fungal and viral infections. Animals that are unable to express Hepcidin due to defects in the USF-2 gene locus are prone to infections from microbes in their environment.
List of References
Acton, RT, Barton, JC & Snively, BM, Geographic and racial/ethnic differences in HFE mutation frequencies in the Hemochromatosis and Iron Overload Screening (HEIRS) Study, The Haematologica Journal, vol. 16, no. 7, pp.16:815-821.
Akarsu, ES & Mamuk, E 2007, Escherichia coli lipopolysaccharides produce serotype- specific hypothermic response in biotelemetered rats, Journal of Physiology Integration, vol. 292, no.5, pp.1846-1850.
Andriopoulos, BT, Corradini, Y & Xia, JL 2009, BMP6 is a key endogenous regulator of Hepcidin expression and iron metabolism, Nature Journals, vol. 41, no. 4, pp. 482-487.
Ashby, DR, Gale, DP & Busbridge M 2007, Erythropoietin administration in humans causes a marked and prolonged reduction in circulating Hepcidin, The Haematologica Journal, vol. 5, no. 8, pp.1-30.
Bothwell, TH 1995, Overview, and mechanisms of iron regulation, Journal of Nutrition Review, vol. 53, no. 37, pp.37.
Bridle, KR, Frazer, DM & Wilkins, SJ 2003, Disrupted Hepcidin regulation in HFE-associated haemochromatosis, and the liver as a regulator of body iron homoeostasis, Lancet Journal, vol. 361, no.14, pp.669-673.
Card, RT 1988, Red cell membrane changes during storage, Journal of Transfusion, vol.2, no.2, pp.40–47.
Collins, H L 2003, The role of iron in infections with intracellular bacteria, Immunol Letter Journal, vol. 85, no.1, pp.193-195.
Corradini, E, Garut, C & Montosi, G 2009, Bone morphogenetic protein signalling is impaired in an HFE knockout mouse model of haemochromatosis, Gastroenterology Journal, vol. 137, no.6 pp. 1489-1497.
Carniel, E 2001, The Yersinia high-pathogenicity island: an iron-uptake island, Microbes Infectious Journal, vol.3, no.4, pp.561-569.
Dallalio, G, Fleury, T & Means, RT 2003, Serum Hepcidin in clinical specimens, Journal of Hematology, vol.122, no.1, pp.1-5.
De Domenico, I 2007, The molecular mechanism of hepcidin-mediated ferroportin down- regulation, Journal of Molecular Biology of the Cell, vol. 18, no.7, pp.2569–2578.
Donovan, A 2000, Nature, Journal of Biology and Chemistry, vol.403, no.8, pp.776-781.
Douglas, SE, Gallant, RS & Liebscher, A 2003, Identification and expression analysis of Hepcidin-like antimicrobial peptides in bony fish, Development Immunology Journal, vol. 27, no.3, pp.589-601.
Faraldo-Gomez, JD & Sansom, MS 2003, Acquisition of siderophores in gram-negative Bacteria, Journal of Current Trends in Microbiology, vol.4, no.7, pp.105-116.
Fillet, G, Beguin, Y & Baldelli, L 1989, Model of reticuloendothelial iron metabolism in humans: Abnormal behaviour in idiopathic hemochromatosis and in inflammation, The Haematologica Journal, vol. 74, no.3, pp.844-851.
Forbes, JR & Gros, P 2001, Divalent-metal transport by NRAMP proteins at the interface of host-pathogen interactions, Journal of Current Trends in Microbiology, vol.9, no.1, pp.397-403.
Gabrio, BW, Finch, CA & Huennekens, FM 1956, Erythrocyte preservation: a topic in molecular biochemistry, Science Journal, vol.11, no.2, pp.103-113.
Gangaidzo, IT, Moyo VM & Mvundura, E 2001, Association of pulmonary tuberculosis with increased dietary iron, Journal of Infectious Diseases, vol. 52, no.87, pp.936-939.
Ganz, T & Nemeth, E 2008, The role of Hepcidin in systemic iron regulation, The New England Journal of Medicine, vol. 357, no.3, pp. 2383-2397.
Ganz, T & Hershiko, F 1978, Body iron loss in animals, Proc Soc Exp Biol Med Journal, no. 159, vol. 12, pp.335-1978.
Goswami, T & Andrew, N C 2006, Hereditary haemochromatosis protein, HFE, interaction with transferrin receptor2 suggests a molecular mechanism for mammalian iron sensing, Journal of Biological Chemistry, vol. 281, no. 39, pp.28494-28498.
Harris, JW & Kellermeyer, RW 1970, The red cell: Production, metabolism, destruction normal and abnormal, Harvard University Press, Cambridge.
Jurado, R L 1997, Iron, infections, and anaemia of inflammation, Journal for Clinical Infectious Diseases, vol.25, no.3, pp.888-895.
Krause, A, Neitz, S & Magert, HJ 2000, A novel highly disulfide-bonded human peptide, exhibits antimicrobial activity, FEBS Lett., vol.480, no.1,pp.147-150.
Lankhors, CE & Wish, JB 2009, role of inflammatory cytokines and Hepcidin, Development Immunology Journal vol.21, no.3, pp.1-25.
Lee, GR 1983, The anaemia of chronic disease, Journal of Seminars in Hematology, vol.20, no.12, pp.61.
Park, CH, Valore, EV & Ganz, T 2001, Hepcidin, a urinary antimicrobial peptide synthesized in the liver, Journal of Biology and Chemistry, vol. 276, no.7, pp.806-7810.
Pietrangelo, A 2004, The role of Hepcidin in systemic iron regulation, The New England Journal of Medicine, vol. 350, no.7, pp. 2383-2397.
Pigeon, C 2001, A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is over expressed during iron overload, Journal of Biology and Chemistry, vol. 276, no.11, pp.7811–7819.
Piperno, A, Girelli, D & Nemeth, E 2007, Blunted Hepcidin response to oral iron challenge in HFE-related hemochromatosism, Journal of Biological Chemistry, vol. 110, no. 12, pp. 4096-4100.
Means, RT & Krantz, SB 1992, Progress in understanding the pathogenesis of the anemia of chronic disease, Journal of Hematology, vol.3, no.24, pp.1639-1647.
Nemeth, E & Ganz, T 2003, Hepcidin, a putative mediator of anaemia of inflammation, is a type II acute phase protein, Clinical Cancer Research Journal, vol. 101, no.1, pp.2461-2463.
Nicholas, GN, Bennoun, M & Devaux, I 2001, Lack of Hepcidin gene expression and severe tissue iron overloads in upstream stimulatory factor two (USF2) knockout mice, Proc National Academic Science Journal, vol. 98, no. 37, pp.8780-8785.
Pigeon, C J, Ilyin, G & Courselaud B 2001, A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is over expressed during iron overload, Journal of Biology and Chemistry, vol.450, no.3, pp.7811-7819.
Sharma, SF, Nemeth, E & Chen, YH 2008, Involvement of Hepcidin in the anaemia of multiple myeloma, Clinical Cancer Research Journal, vol. 14, no. 11, pp. 3262-3267.
Waheed, AT, Parkkila, AS & Saarnio, J 1999, Association of HFE protein with transferrin receptor in crypt enterocytes of human duodenum, Proc National Academic Science Journal, vol.96, no.1, pp.1579.
Weinberg, ED 2000, Microbial pathogens with impaired ability to acquire host iron, Journal of Transfusion, vol.13, no 1, pp.85–89.
Weinstein, DA, Roy, CN & Fleming, MD 2002, Inappropriate expression of Hepcidin is associated with iron refractory anemia: Implications for the anemia of chronic disease, Journal of Hematology, vol. 100, no.2, pp.3776-3781.
Winkelmann, G 2002, Microbial siderophore-mediated transport, Journal Biochemical Society Transactions, vol.30, no.5, pp.691-696.
Xia, JL, Babitt, Y & Sidis, R 2008, Hemojuvelin regulates Hepcidin expression via a selective subset of BMP ligands and receptors independently of neogenin, Journal of Infectious Diseases, vol. 111, no.10, pp. 5195–5204.
Yang, D, Chertov, O & Bykovskaia, S N 1999, Hepcidin, Science Journal, vol. 286, no. 5, pp.525–528.