Modern medicine achievements have reached a level that allows for treatment for multiple diseases threatening the lives of mankind. However, there still remain a number of ailments the cure for which has not been discovered. Among those, a group of brain diseases is recognized, occurring both in humans and animals and generally called prion diseases. The first public interest in such diseases arose in the mid-1980s when there was an epidemic of bovine spongiform encephalopathy (BSE) affecting cattle in the United Kingdom. Consequently, concern over whether BSE could be passed on to humans grew and waste research on prion diseases was being carried out in world laboratories. According to up-to-date medical data, prion diseases are transmissible and render a spongy appearance to the brain tissue due to its destruction; for those reasons such diseases are also called transmissible spongiform encephalopathies or TSEs, the typical signs of which are loss of coordination and — in humans — dementia. There exists a number of diseases falling into the category of TSEs, among them being: Creutzfeldt–Jakob Disease, variant Creutzfeldt–Jakob Disease, Bovine Spongiform Encephalopathy, Kuru, Gerstmann-Sträussler–Scheinker disease, Fatal Familial Insomnia, Scrapie and some other animal TSEs (Kimball).
A more widespread form of prion diseases in humans, Creutzfeld–Jakob disease (CJD), was first described much earlier than the BSE epidemic occurred, in 1920, by a German neurologist Hans Gerhard Creutzfeldt, and a bit later by his colleague Alfons Maria Jakob, thus acquiring a double name. Being extremely difficult to diagnose, Creutzfeld–Jakob disease is annually reported to affect on average one person per million worldwide. Thus, it appears of vital importance to establish the causes and the pathophysiological features of this disease in order to be able to trace its development in an attempt to find a cure for it.
In their attempts to find the cause and essence of Creutzfeld-Jakob disease, scientists noticed that transmission of CJD occurred during injections of brain tissue from an animal or human patient with a prion disease into another animal. This suggested the causative factor for the CJD to be an infectious agent such as a virus. But despite multiple efforts, including treatment of the “infected” tissues with heat and ultraviolet (which still did not reduce their infectiousness), no evidence of the viral nature of CJD was found. Only in the 1980s the current director of the Institute for Neurodegenerative Diseases at the University of California, San Francisco, and a 1997 Nobel Prize Laureate, Stanley Prusiner, discovered and pioneered the research of prions, a class of infectious self-reproducing pathogens primarily or solely composed of protein, and claimed them to be the cause of Bovine Spongiform Encephalopathy (“mad cow disease”) and its human equivalent, Creutzfeldt–Jakob disease. Thus, originally considered to be a virus infection, Creutzfeld–Jakob disease is now recognized to be the consequence of an abnormal form of a specific protein called a prion (shortened form for proteinaceous infectious particle).
In order to efficiently understand the mechanism of prion diseases, it is essential to trace how they originate. A major and crucial role in the development and functioning of the human body is played by proteins — “molecules made up of thousands of smaller chemical units called amino acids, joined together like beads on a necklace” (Creutzfeld–Jakob Disease Foundation 7). Being flexible, protein molecules can adopt a number of shapes — a quality that is decisive for the development of prion diseases. As such, prion protein molecules are normal cellular proteins present in many organs and tissues, including the brain, spinal cord, and eyes of healthy humans and animals. Compared to normal body protein molecules, prion proteins appear to have changed their three-dimensional configuration (Kimball).
According to Prusiner, there are two possible forms of the prion protein:
“The normal isoform of prion protein (PrPc) is protease-sensitive and is expressed in many tissues, but chiefly in neurons. Protease resistant form of this protein, found in diseased brains is designated PrPsc where ‘sc’ stands for ‘Scrapie isoform’.” (qtd. in Prabhakar and Bhatia 325)
The abnormal prions, PrPsc, possess a number of characteristics that distinguish them from the normal ones: firstly, they are not broken down by enzymes, and secondly, they are prone to forming tiny fibers called scrapie associated fibrils (SAFs) — highly infectious tissues which, when congregated, make up a chemical structure called amyloid. As autopsy examinations have shown, amyloid deposits are frequent in the brain of patients who have fallen victims to Creutzfeld–Jakob disease (Creutzfeld–Jakob Disease Foundation 7). Primarily, the PrPsc structure reminds that of the PrPc, but the secondary structure of PrPsc is dominated by beta conformation. Moreover, PrPsc is almost non-solvent and are highly resistant to digestion by proteases, which makes them quite impossible to destroy (Kimball).
To make matters worse, Prusiner discovered the dangerous nature of PrPsc reveals itself in the way PrPsc molecules behave when encountering the normal PrPc molecules:
“… a single molecule of PrPSc can convert molecules of PrPC into the abnormal form. These newly converted molecules can in turn “corrupt” more normal molecules leading to a cascade effect which would eventually cause brain damage.” (Creutzfeld–Jakob Disease Foundation 8)
Thus, the abnormal prion functions as an infectious agent due to its ability to convert normal prions into abnormal forms. The interaction of the normal and the abnormal prions leads to a dramatic increase in the latter amount consequentially causing a plaque in the brain. The mechanism of encounter is illustrated in the following scheme:
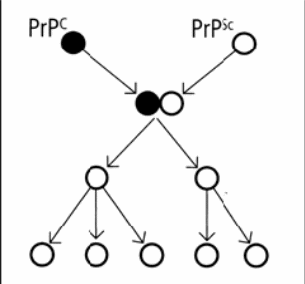
The danger of the PrPsc, therefore, consists in its ability to promote refolding of native PrPc proteins into the diseased state. This process occurs in two steps: firstly, the alpha-helices are unfolded; and secondly, the matter refolds to beta-pleated sheets. As a result of the exponential increase in the misfolded protein molecules, a large number of insoluble prions is accumulated in the affected cells disrupting cell membrane function and causing cell death. Being practically indigestible, Proc proteins are produced in a kind of self-sustaining feedback loop, spreading and multiplying at an extremely rapid rate and in most cases leading to the death of the patient within a couple of months.
Bearing all the aforesaid in mind, it is all the more essential to understand the origins of Creutzfeld–Jakob disease in the body and to trace the ways it is transmitted, i.e. the ways the PrPsc prions appear in the affected tissues. In this case, scientists single out several options of possible CJD transmission, depending on the type of disease in each specific instance. The most widespread occurrence of Creutzfeld–Jakob disease observed (around eighty – eighty-five percent of cases) is the so-called sporadic, or spontaneous CJD, the cause of which is still claimed to be unknown (Prabhakar and Bhatia 325). This type of CJD affects mostly patients 55–65 years of age and is characterized by a short course during which ataxia and dementia are observed.
Another ten to fifteen percent of cases are the familial CJD, caused by an inherited mutation of the PrP gene. This type of disease is passed on from parent to child at conception through the DNA and can be usually recognized from a family history of the illness in brothers, sisters, or parents. There can be occurrences when the disease is not passed on: each child born from a parent carrying genetic CJD has a fifty percent chance of inheriting the disease-causing mutation. As compared to sporadic CJD, familial CJD usually starts at an earlier age and lasts longer.
The final, third type of CJD is acquired CJD, responsible for about five percent of all cases. It comprises two subtypes: a) the iatrogenic CJD, occurring when a person is contaminated through brain surgery, corneal transplant, dura mater graft, or growth hormone (here the contamination risk is extremely high when instruments are applied which had previously been used on a CJD-contaminated person; even after two years they still bear the infectious matter; thus nowadays instruments which have been used on the brain of someone with suspected CJD are destroyed); and b) the variant CJD, which starts as a result of exposure to BSE through consumption of infected beef or blood or plasma transfusion. Variant CJD was first identified in 1995 in two teenagers and since then has attracted specific attention due to peculiarities of its occurrence: much earlier age of the affected people and a potential epidemic risk caused serious legislative measures to be taken about cattle-based foods in the human food supply. Action has been taken since 1989 to remove those parts of cattle where the greatest concentrations of an infective agent are found, including brains and spinal cords, from the human food chain.
All-in-all, a heated debate over the actual cause of prion diseases is going on, with prion being ascribed status of the agent which causes disease or a mere symptom caused by a different agent. In 2007 a Yale University neurologist, Laura Manuelidis, claimed to have found a virus-like particle (without finding nucleic acids so far) in less than ten percent of the cells of a scrapie-infected cell line and a mouse cell line infected by a human CJD agent. However, Prusiner’s protein-only hypothesis is so far mostly based on the evidence thus holding a leading position in the explanation of the prion diseases cause.
With new variants of Creutzfield-Jakob disease being discovered, and the threat of its epidemic impending the world unless duly managed, it becomes one of the significant public health issues to properly establish the true causes and pathology of the disease so that a true and efficient cure could be developed for providing patients with hope for convalescence and relieving the menace of contamination from the healthy part of the population.
Works Cited
Creutzfeld–Jakob Disease Foundation. Creutzfeld-Jakob Disease and Other Prion Diseases. 4th ed. Akron, OH: Creutzfeldt-Jakob Disease Foundation, Inc., 2009.
Prabhakar, Sudesh and Rajinder Bhatia. “Diagnosis of Creutzfeldt–Jakob Disease.” Neurol India 49.4 (2001): 325-8.
“Prion Diseases.” Kimball, John W. Kimball’s Biology Pages. 2009. Web.