Introduction
Genotyping refers to the process of finding differences in the genetic constitution of an individual through the examination of the entity’s DNA sequence using biological assays. The DNA sequence is then compared to a reference sequence to pinpoint the disparities (Abdurakhmonov 2018). Such a process may uncover alleles that have been received from parents as well as genetic modifications that can result in key phenotypic changes, for example, differences that contribute to the uniqueness of an individual and pathological alterations caused by disease. Genotyping has numerous applications in medicine, fundamental scientific investigations, and agriculture. Different molecular markers are used as the basis of identification of genetic differences within populations, including single-nucleotide polymorphisms (SNPs).
SNPs are changes in a single base-pair in DNA that ensue at precise loci within the genome. For instance, most individuals have the cytosine (C) nucleotide at a particular base locus in the genome. However, in a small section of individuals, this base is substituted by an adenine (A). This phenomenon implies that a SNP exists at this position with two probable nucleotide variations of C or A. The human genome has more than 660 million SNPs, making them the most predominant genetic variation (van Arensbergen et al. 2019).
These SNPs are associated with many physical traits, for example, skin and eye colour as well as hereditary diseases such as sickle cell anaemia and cystic fibrosis (Li et al. 2019). The presence of SNPs can also act as indicators of the probability of developing certain diseases such as Alzheimer’s disease, certain forms of cancers and diabetes.
SNP rs2234693 is found on the oestrogen receptor (ER) 1 gene, which is located on chromosome 6 of the human gene (Liu et al. 2018). This SNP entails a change in the nucleotide thymine (T) that is substituted with C. The ER gene has two variants: alpha (α) and beta (β). ER-α serves in facilitating hormonal responses in tissues that are sensitive to oestrogen, such as the breasts, skin, hair, urinary and reproductive tract (Perkins, Louw-du Toit & Africander, 2017).
It executes these functions with the aid of different domains such as those responsible for the regulation of hormone production, initiation of transcription and binding of DNA. There is sufficient evidence to implicate oestrogen receptor-α as the key receptor responsible for the initiation of breast cancer (Tian & Schiemann 2017). SNP genotyping has transformed personalised medicine by envisaging a person’s risk of acquiring certain illnesses or formulating targeted therapies that correspond to the genetic origin of the disease. SNPs can also predict an individual’s response to treatment, thereby guiding the selection of the best course of treatment.
New and known SNPs can be detected using different methods such as DNA sequencing, SNP microarrays, mass spectrometry and techniques that rely on polymerase chain reaction (PCR). Overall, SNP detection methods can be classified as either discovery or screening assays. SNP discovery involves the uncovering of unknown SNPs in specific areas or the entire genome. In contrast, SNP screening entails the classification of individuals (genotyping) based on the presence or absence of specific SNPs or determining the association between particular SNPs and known traits (Liu et al. 2018; Perkins, Louw-du Toit & Africander, 2017).
PCR is a molecular biology technique that enables the production of millions of copies of DNA in-vitro. It depends on the activity of the enzyme Taq polymerase and short DNA primers that are complementary to the sequence to be amplified (Kadri 2019). Its advantages include accuracy, ability to be scaled to high throughput, and ease of performance. The bioinformatic analysis involved in real-time PCR is less complex than other methods such as microarrays and sequencing (Yu et al. 2017).
Specific SNPs in target sequences can be detected using TaqMan 5′-nuclease chemistry (Beltz et al. 2018). In the absence of real-time PCR technology, conventional PCR can be used to identify SNPs in DNA. The main shortcoming of conventional PCR is the need for gel electrophoresis to separate the amplified DNA fragments, which lengthens the process. Conventional PCR is often used alongside restricted fragment length polymorphisms (RFLP) in SNP diagnosis (Liu et al. 2018).
RFLP is a molecular biology technique that takes advantage of differences in the DNA sequence of homologous DNA (polymorphisms) to tell between individuals or populations. It also refers to differences in the cutting sites of restriction endonucleases within a given DNA sequence. During an RFLP assay, a piece of DNA is cut into fragments using one or more restriction enzymes. The resultant restriction fragments are separated by gel electrophoresis based on their sizes. The final pattern of the separated fragments is analysed to determine any differences.
SNPs can generate or eliminate restriction enzyme recognition sites, thereby changing the number and size of DNA fragments obtained from digestion with restriction endonucleases. The main shortcoming of RFLP is that isolating sufficient quantities of DNA for the assay is labour intensive and time-consuming. Nonetheless, this problem can be circumvented by combining PCR with RFLP (Yu et al. 2017). PCR facilitates the amplification of small quantities of DNA for subsequent RFLP analysis, making it possible to analyse many samples in a short time. However, for the entire assay to be successful, precise reaction conditions and concentrations should be determined. The purpose of this report is to develop and optimise a PCR-RFLP based genotyping assay to identify SNPs associated with disease on the human oestrogen receptor 1 gene.
Methods
Materials
The restriction enzyme used for the assay was the PvuII Fast Digest enzyme from Thermo Fisher Scientific. The PCR buffer provided was 10x Green Buffer from the same manufacturer. Other materials that were provided included reverse and forward Primers, Taq Polymerase, dNTPs, PCR Buffer, known DNA samples (heterozygous, homozygous mutant and homozygous wildtype) and an unknown DNA sample.
Marker Selection
The DNA sequence of the ER1 gene was retrieved from the NCBI website. Primer-BLAST was then used to identify the forward and reverse primer sequences, which were 5’-TCACACATCACCATTCTCAGC-3’ and 5’CTTTCATTACCTCTTGCCGTC-3’. The suggested annealing temperature was 57oC. PvuII was the ideal restriction enzyme because it covers the region of the SNP rs2234693.
Gradient Polymerase Chain Reaction
A gradient PCR reaction was conducted to optimise the amplification conditions for the assay. The total reaction mixture was 25µl, which consisted of 3µl template DNA and 22 µl of other reagents. The PCR master mix comprised 2.5µl buffer, 0.5µl of dNTPs, 0.5µl forward primer, 0.5µl reverse primer, 0.125 units/µl of Taq polymerase, 3µl of template DNA and 17.875µl of nuclease-free water to make up the rest of the volume.
Ten PCR Eppendorf tubes were filled with 22µl of the master mix followed by the addition of 3µl of template DNA at a concentration of 70ng or water for the negative control. The PCR reaction mixture was transferred to a thermocycler for a gradient PCR reaction. The primer annealing temperature was optimised from 54 ̊C to 60 ̊C. The conditions were set to be followed in the following order: initial DNA denaturation for 3 minutes at 94 ̊C, 30 cycles of extension consisting of denaturation at 95 ̊C for 1 minute, primer annealing at the gradient temperatures (52 ̊C to 61 ̊C) for 1 minute, DNA extension at 72 ̊C for 1 minute, with a final 10-minute incubation at 72 ̊C.
Gel Electrophoresis Analysis of PCR Products and RFLP Analysis
The PCR products were subjected to agarose gel electrophoresis to visualise them. A 1.5% agarose gel was prepared by dissolving 0.52g of agarose powder in 35ml of 1x TBE buffer. The mixture was heated in a microwave at 30-second intervals for 3 minutes to dissolve the powder and avoid overboiling the mixture. The molten gel was cooled to 55°C followed by the addition of 0.3μl of Gel Red. The mixture was poured into a gel tray fitted with endplates and a well comb and allowed to solidify. Enough 1x TBE buffer to cover the gel was added to the electrophoretic tray. DNA was loaded into the wells after which the gel was run at Run the gel at a voltage of 80 mA for 30 minutes. The gel was then analysed in a gel documentation chamber. The optimum annealing temperature was determined from the resultant bands.
The PCR products were then subjected to restriction enzyme digestion and gel electrophoresis. The total restriction digest volume was 25µl and consisted of 16µl of nuclease-free water, 1µl of PvuII, 3µl of 10x FastDigest Green Buffer, 10µl of the PCR product and bromophenol blue dye. The mix was incubated at 37oC. A DNA marker and the digestion products were loaded onto their respective wells in a 1.5% agarose gel. The products were electrophoresed, and the gel was then analysed by a gel documentation chamber.
Results
The gradient PCR showed that the optimum annealing temperature for the primer pairs was 58oC. PCR products from this run were used in the subsequent restriction digestion. Different banding patterns were produced when the PCR products were digested with PvuII. Figure 1 shows the gel electrophoresis of the PCR products. Very faint bands were seen for only T1 and T0.
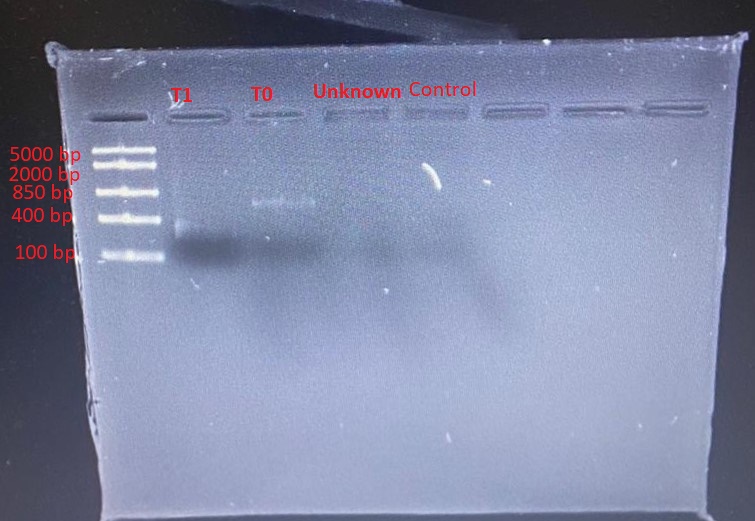
Discussion
The aim of the experiment was to develop and optimise an assay to identify different genotypes of the ESR1 gene based on the SNP rs2234693. The three components of the assay that were crucial to its success were PCR amplification, restriction digestion and gel electrophoresis. A PCR assay involves three main steps of denaturation, primer annealing, and extension, which are carried out at different temperatures.
The cycle is repeated about 30 times to produce adequate copies of the template DNA. This process is done in specialised equipment known as a thermocycler. Real-time PCR, which is also referred to as quantitative PCR, entails the simultaneous amplification and quantification of specific DNA sequences during the reaction. It enables the screening of known SNPs. Optimising a PCR assay entails determining the correct primers to ensure that the target DNA sequence is amplified.
The chosen primer pair was selected based on its ability to span the target sequence adequately as reported by Ramesh et al. (2020). The primer should anneal properly to its complementary sequence before Taq polymerase can extend the primer and synthesise the new strand. The gradient PCR run showed that the optimum annealing temperature was 58oC.
An accurate RFLP assay should use a suitable restriction enzyme. Many studies to identify SNP rs2234693 have used the restriction enzyme PvuII (Pontin et al. 2018; Yin et al. 2018; Ahrar et al. 2019). The findings from this experiment also showed that the enzyme produced different banding patterns on the PCR products.
Gel electrophoresis enables the separation of DNA fragments by size in agarose gel through the application of an electric field. DNA is a negatively charged biomolecule. Therefore, it migrates from the negative electrode to the positive electrode under the influence of an electric field. Several factors determine the efficiency of an electrophoretic run for DNA separation, including agarose gel concentration, the choice of buffer, voltage and duration of the electrophoretic run, DNA concentration and gel staining (Lee et al. 2012).
Figure 1 shows very faint DNA bands in lanes 2 and 3, whereas lanes 4 and 5 showed no band. This problem could be attributed to an insufficient amount of DNA loaded, inadequate staining, diffusion of DNA into the gel or masking of DNA by the tracking dye (Green & Sambrook 2019). However, given that the molecular ladder bands were distinct, the plausible explanation for the faint bands was a low concentration of DNA. There was a likelihood that the DNA was too dilute.
This problem could be solved by increasing the concentration of DNA. It was also possible that the amount of DNA loaded was lower than the ideal concentration for the size of wells used. The recommended loading concentration is approximately 0.1 to 0.2 µg per 1 mm of gel lane width. However, the fact that lane 5 (negative control) had no band indicated the validity of the assay because no DNA was included in the PCR mix for the negative control.
The gel should also be imaged as soon as possible because prolonged storage could cause the diffusion of DNA fragments, leading to low band intensity. Part of the DNA could have run off the gel. To avoid this problem, it was necessary to ensure that the gel was immersed in the buffer. The problem of DNA masking by tracking dyes could be avoided by ensuring that the correct amount of tracking dye used during sample preparation.
Conclusion
From the experiment, it was concluded that PCR-RFLP is a reliable method of determining SNP rs2234693 on the human ER1 gene. The optimum annealing temperature is 58oC. However, more trials are needed to optimise the gel electrophoresis of the PCR products to produce distinct bands.
Reference List
Abdurakhmonov, I.Y. (ed.) (2018) Genotyping. London: IntechOpen.
Ahrar, H., Aghili, K., Sobhan, M.R., Mahdinezhad-Yazdi, M., Akbarian-Bafghi, M.J., and Neamatzadeh, H. (2019) ‘Association of rs2234693 and rs9340799 Polymorphisms of Estrogen Receptor-1 Gene with Radiographic Defined Knee Osteoarthritis: A Meta-Analysis.’ Journal of Orthopaedics 16(3), 234-240.
Beltz, K., Tsang, D., Wang, J., Rose, S., Bao, Y., Wang, Y., Larkin, K., Rupp, S., Schrepfer, D., Datta, K., and Gunderson, K. (2018) ‘A High-Performing and Cost-Effective SNP Genotyping Method Using rhPCR and Universal Reporters.’ Advances in Bioscience and Biotechnology 9(09), 1-16.
Green, M.R. and Sambrook, J. (2019) ‘Analysis of DNA by Agarose Gel Electrophoresis.’ Cold Spring Harbor Protocols, 2019(1), 87-94.
Kadri, K 2019, Polymerase Chain Reaction (PCR): Principle and Applications. Web.
Lee, P.Y., Costumbrado, J., Hsu, C.Y., and Kim, Y.H. (2012) ‘Agarose Gel Electrophoresis for the Separation of DNA Fragments.’ JoVE (Journal of Visualized Experiments) 62, 1-5.
Liu, X., Huang, J., Lin, H., Xiong, L., Ma, Y., and Lao, H. (2018) ‘ESR1 PvuII (rs2234693 T> C) Polymorphism and Cancer Susceptibility: Evidence from 80 Studies.’ Journal of Cancer 9(16), 2963-2972.
Perkins, M.S., Louw-du Toit, R., and Africander, D. (2017) ‘A Comparative Characterization of Estrogens Used in Hormone Therapy via Estrogen Receptor (ER)-α and-β.’ The Journal of Steroid Biochemistry and Molecular Biology 174, 27-39.
Pontin, P.A., Nogara, P.R.B., Fonseca, F.C.P., Netto, C.C., Carvalho, K.C., Junior, J.S., Baracat, E.C., Fernandes, T.D., Maffulli, N., Santos, M.C.L., and Godoy-Santos, A.L. (2018) ‘ERα PvuII and XbaI Polymorphisms in Postmenopausal Women with Posterior Tibial Tendon Dysfunction: A Case Control Study.’ Journal of Orthopaedic Surgery and Research 13(1), 1-5.
Ramesh, S.S., Christopher, R., Devi, B.I., Bhat, D.I., and Shukla, D (2020) ‘Estrogen Receptor Alpha Gene Variant, Pvuii (Rs2234693), As a Potential Pharmacogenetic Biomarker for Aneurysmal Subarachnoid Hemorrhage in Postmenopausal Women.’ The Pharmacogenomics Journal 1-9.
Tian, M. and Schiemann, W.P. (2017) ‘TGF-β Stimulation of EMT Programs Elicits Non-Genomic ER-Α Activity and Anti-Estrogen Resistance in Breast Cancer Cells.’ Journal of Cancer Metastasis and Treatment 3, 150-160.
van Arensbergen, J., Pagie, L., FitzPatrick, V.D., de Haas, M., Baltissen, M.P., Comoglio, F., van der Weide, R.H., Teunissen, H., Võsa, U., Franke, L., and de Wit, E. (2019) ‘High-Throughput Identification of Human SNPs Affecting Regulatory Element Activity.’ Nature Genetics 51(7), 1160-1169.
Yin, X.Q., Ju, H.M., Guo, Q., Zhao, L., Zhu, X.X., Wei, R., Zhang, Z., Zhang, Y.H., Wang, B., and Li, X. (2018) ‘Association of Estrogen Receptor 1 Genetic Polymorphisms with Recurrent Spontaneous Abortion Risk.’ Chinese Medical Journal 131(15), 1857.
Yu, H., Zheng, M., Zhang, L., Li, H., Zhu, Y., Cheng, L., Li, L., Deng, B., Kijlstra, A., and Yang, P. (2017) ‘Identification of Susceptibility SNPs in IL10 and IL23R-IL12RB2 for Behçet’s Disease in Han Chinese.’ Journal of Allergy and Clinical Immunology 139(2), 621-627.