Introduction
Technological advances are crucial for the development of healthcare around the world, as they help to improve treatment and diagnosis methods of various conditions. According to a recently published article in Forbes magazine, there are at least nine current trends in technology that will transform medicine and healthcare in the nearest future (Marr). These nine trends include AI and machine learning, robotics, computer and machine vision, wearable tech, genomics, 3D printing, extended reality, digital twins, and 5G (Marr). While some of the technologies are used only by high-level professionals in very specific scenarios, others are available to the public for every-day use. For instance, the utilization of wearable medical monitoring devices has become a trend in recent years.
Wearable fitness and medical monitoring technology has weaved itself into society so that smartwatches, fitness trackers, and other monitoring devices are now mainstream. Medical wearable technology includes devices that a consumer can wear to record data of a user’s health metrics and exercise (Phaneuf). Wearable devices include fitness trackers, smartwatches, electrocardiogram (ECG) monitors, blood pressure monitors, and biosensors. The use of such devices jumped to 33% in 2018, from only 9% in 2014 (Phaneuf). Thus, it can be stated that wearable medical devices have become a matter of increased interest to the public and healthcare professionals.
Wearable devices are mostly used to measure heart-related metrics, such as blood pressure, pulse, and ECG. This implies that wearable devices can be used to diagnose and address cardiovascular diseases. Cardiovascular diseases cause almost 18 million deaths every year worldwide (World Health Organization). In other words, more than 31% of all deaths are attributed to heart-related conditions, which makes them the number one cause of death around the globe (World Health Organization). In the US, heart diseases are associated with 25% of deaths and $219 billion of healthcare costs (Centers for Disease Control and Prevention, “Heart Disease”). However, cardiovascular diseases are known to be preventable, and wearable technology can help the society to address the problem.
Wearable devices, however, need to be of sufficient quality and satisfy several regulations to be distributed among the consumers. These regulations may be different depending on the region of distribution. The present paper focuses on the likely regulatory classification and approval pathway for QardioArm, a wearable blood pressure monitor. The paper offers a thorough description of the product, its regulatory pathway in the US, and a comparison of these requirements with the EU region.
Product Background
Description of the Technology
QardioArm is a wearable wireless blood pressure monitor produced by Qardio, a US-based company. The device is marketed as a smart blood pressure monitor, as it is used in combination with a smartphone. The device is a small plastic box with a band that can be put around a person’s arm to measure blood pressure and heart rate. QardioArm is launched using an application run on a smartphone, tablet, or Kindle (Qardio). The medical device is currently compatible with iOS 10.0 or later, Kindle, Android 5 or later, and Apple WatchOS 3 (Qardio). QardioArm uses Bluetooth 4 technology to connect with other devices. In summary, QardioArm is a relatively small medical device that can be used with a wide variety of smart devices to measure blood pressure and heart rate.
QardioArm is easy to use and simple to set up that provides insightful information for the user. In order to start measuring blood pressure, a person needs to put the device around the upper part of the arm, download the application to a smartphone, tablet, or Kindle, connect the devices via Bluetooth, and press the Start button in the application (Qardio). The results are automatically displayed on the screen and stored in the app (Qardio). The measurements are insightful even for almost every user, as the results are automatically interpreted against the World Health Organization blood pressure chart (Qardio). All the features described above are helpful for uneducated users to track their well-being.
QardioArm gives the user an ability to take precise measurements that were validated in clinical trials. The device has the feature to measure blood pressure three times in a row (triple measurement technology) and display the average of these three measurements, which helps to avoid erroneous data (Qardio). A study of 100 healthy volunteers conducted by Pardo et al. revealed that QardioArm displayed consistent readings within and across sessions (e198). The study confirmed that QardioArm adheres to the standards set by the European Society of Hypertension International Protocol (ESH-IP). These results were confirmed by Chahine et al. on a sample of 33 subjects (18). In summary, the QardioArm device and application can be used without fear of erroneous measurements.
The mobile application provides the user with the ability to store, monitor, and share information about blood pressure and heart rate. After taking measurements, the data is automatically recorded to the application on the user’s smartphone with the association to location acquired from GPS (Qardio). It can be referenced or deleted at any time without complications (Qardio). The history of measurements is presented in the form of a chart, which helps to understand the dynamics of blood pressure and heart rate at a set period (Qardio). Moreover, the data can be shared with family members or other stakeholders via Bluetooth or the Internet (Qardio). The application also has several features that enhance QardioArm’s usability. The useful features include making notes, setting goals and reminders, and tracking irregularities in measurements. In other words, the device makes the data about blood pressure and heart rate easily accessible and comprehensive.
The weight of the devices is 0.68 pounds, and its dimensions are 5.5 x 2.7 x 1.5 in (Qardio). The cuff size is flexible between 8.7 in and 14.6 inches in circumference (Qardio). The device uses the oscillometric method of measurement with automatic inflation and a controlled pressure release valve (Qardio). The method uses a closed air pipe system to sense the vibrating signal (Food and Drug Administration 2, ”Ki140067”). The microcomputer automatically detects the characteristics of the pulse signal, which is different from the traditional method based on Korotkov sound (Food and Drug Administration 2, “Ki140067”). The method is associated with high accuracy and consistency. The range of measurements is 40-250 mm Hg for blood pressure and 40-200 beats/min for pulse (Qardio). The resolution of the measurements is 1mm Hg for blood pressure and 1 beat/min for heart rate (Qardio). QardioArm uses four AAA batteries as a power source (Qardio). In short, QuardioArm is a small high-precision wearable device used to measure blood pressure and pulse with high resolution and sensitivity.
User and User Environment
The device is used by the patient to control blood pressure at any convenient time and place. Qardio is intended for personal use by the patient and is not considered a standard tool for measuring blood pressure and heart rate in clinical settings (Pardo et al. e198). It can be used for reference only, and doctors cannot rely on the device as a diagnostic tool. However, healthcare providers can benefit from consulting the history of measurements provided by the patient. In other words, doctors can use the measurements as a screening tool. Patients, however, can use the device at any place and time as long as they follow measurement recommendations. The manufacturer advises that the patient needs to put the cuff on the left arm at the level of the heart, sit comfortably, and rest the arm on a table or any other surface (Qardio Support). Patients can use the device without following the recommendations; however, it can lead to inaccurate results.
Intended Use
The devices can be used by anyone who wishes to control his or her blood pressure and heartbeat. However, the device is intended for patients with elevated blood pressure and hypertension. According to the Centers for Disease Control and Prevention, 29% or almost 75 million Americans, both males and females, have hypertension (“Facts about Hypertension”). In their study, Zhang et al. revealed that using wearable measurement devices to monitor blood pressure is associated with increased compliance with hypertension recommendations. Consistent blood pressure monitoring is essential for evaluating how effective the treatment for hypertension is. Without proper treatment and regular monitoring, hypertension may lead to coronary heart disease, stroke, or death (Centers for Disease Control and Prevention, “Facts about Hypertension”). Therefore, the indications for the use of the device is elevated blood pressure and treatment for hypertension.
Contraindications and Warnings
While almost anyone can use QardioArm, users need to be aware of contraindications and warnings about the utilization of the device. In particular, QardioArm is not recommended for people with serious asthma (“QardioArm User Manual”). Moreover, cuff inflation may cause internal bleeding in people with severe blood flow problems and blood disorders (“QardioArm User Manual”). Therefore, it is recommended that patients with such problems contact a doctor before using blood pressure monitors.
Among other warnings, the user manual says that common arrhythmias, ventricular premature beats, atrial fibrillation, arterial sclerosis, poor perfusion, diabetes, age, pregnancy, preeclampsia or renal disease can affect the performance of the automated sphygmomanometer or its blood pressure reading (“QardioArm User Manual”). The device is not intended for measuring the frequency of heart pacemakers (“QardioArm User Manual”). In summary, QardioArm has few contraindications and warnings, which implies that it can be used by the majority of people without fear of injury or damage to health.
Regulatory Pathway
Possible Pathways
In the US, the Food and Drug Administration (FDA) is responsible for the efficacy and safety of medical devices and drugs. There are several pathways a new device can be approved to the market. In general, before marketing a new device, a manufacturer needs to understand if a new product is a device, understand the intended population that will use it, classify the device, clinically test the device if needed, and submit an application form to the FDA (Van Norman 282). The pathway, however, differs for various devices depending on their classification and intended use.
There are three classes of medical devices that differentiate them according to associated risks. Class I devices have a low risk of illness or injury, while Class II devices are associated with moderate risks (Van Norman 278). Devices that are of unreasonable risk of harm aim at preventing impairment of human health, or help to sustain human life are Class III devices (Van Norman 278). Class I and II devices do not undergo a rigorous review and require minor or no clinical trials, while Class III devices require significant testing and evaluation (Van Norman 278). The classification also determines which pathway a device needs to follow pre-market approval (PMA), pre-market notification (PMN), or humanitarian device exemption (HDE) pathways.
The PMA process is the most rigorous pathway to marketing a new device. PMA is applied to all devices that do not have an existing analog that can be used as a reverence (Van Norman 279). In order to be approved through PMA, a device needs to acquire Level I or II evidence of the device’s efficacy and safety (Van Norman 279). Clinical trials are allowed only when the device has acquired an investigational device exemption (IDE). After the trials are completed, the sponsor of the device needs to submit a PMA form, which is reviewed by the Center for Devices and Radiological Health (CDRH). CDRH has up to 180 days to accept or reject the application.
PMN, which is also known as the 510(k) application, is used when a new device has an existing predicate device on the market. Such devices are rarely required to go through clinical trials and approved if there is enough evidence that they are similar to an existing analog (Van Norman 281). This process is critiqued by the public, as it creates incentives for manufacturers to make only slight changes to existing devices and charge a premium price as if it is a new device (Van Norman 281). The application is also reviewed by the CDRH, which needs to make a decision within 60 days (Van Norman 282). In short, PMN is a much faster and simpler process in comparison with PMA.
The third pathway, HDE, is meant for humanitarian use devices (HUDs) that are expected to treat or diagnose less than 4,000 US citizens (Van Norman 283). The HDE process is similar to PMA; however, scientific evidence of efficacy is not required (Van Norman 283). However, significant evidence of safety is still required. The decision about the device is given within 45 days from filing the documents (Van Norman 283). An investigational device may also be used under emergency and expanded approvals to save the life of a patient if no alternative methods are present. All the possible pathways are demonstrated in Figure 1 below.
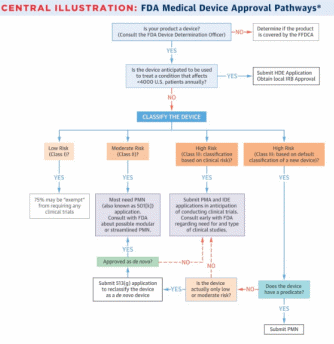
As can be seen from above, the choice of a regulatory pathway may become a challenging task. Therefore, sponsors of devices are recommended to have early consultations with the FDA through pre-submission meetings (Van Norman 284). These meetings can help to classify the new device, decide on the level of clinical evidence needed for approval, and fill in the required forms.
QardioArm’s Pathway
QardioArm was approved by the FDA through PMN as it had substantially equivalent to a pre-existing device. The FDA received the PMN form for QardioArm on January 10, 2014 (Food and Drug Administration, “510(k)”). The device was classified as a Class II product as per 21CFR870.1130, which stands for non-invasive blood pressure measurement systems, and its 510(k) number was K140067 (Food and Drug Administration, “510(k)”). The device is meant to measure systolic, diastolic, mean, or any combination of the three pressures that can be measured from the use of transducers on the surface of the body (Food and Drug Administration, “510(k)”). In order for the FDA to approve QardioArm, the sponsor needed to prove that the device was substantially equivalent to a predicate device.
QardioArm was determined to have at least two predicate devices, which were KD-936 Fully Automatic Wireless Blood Pressure Monitor (K120672) and Upper Arm Blood Pressure Monitor, model BP-700NW and Bluetooth Transmission BP-700W (K121025) (Food and Drug Administration 2, “Ki140067”). FDA’s analysis revealed that the only difference between QardioArm and predicate devices was the visual appearance, interface, and cuff size, while other parameters were touched insignificantly (Food and Drug Administration 2, “Ki140067”). Therefore, the device did not need to pass through any clinical trials, as it was essentially the same as devices already on the market.
As the device was found not eligible for PMA, the sponsor could start marketing the product, which was subject to the general controls provisions of the Federal Food, Drug, and Cosmetic Act. However, the permission to market the device did not mean that QardioArm complied with the Act; instead, the manufacturer needed to make sure that the device complies with a number of requirements. These requirements included registration and listing (21 CFR Part 807), labeling (21 CFR Part 801), medical device reporting (21 CFR Part 803), and quality systems (QS) regulation (21 CFR Part 820) (Food and Drug Administration 3, “Ki140067”).
Before officially marketing a product, it needed to be registered using the FDA Unified Registration and Listing System (Food and Drug Administration, “21CFR801”). The registration information needs to include details about the manufacturer and the medical device it produces, including the PMN number (Food and Drug Administration, “21CFR801”). The information needs to be confirmed annually from October 1st to December 31st.
The QS regulations describe the requirements for the entire medical device lifecycle, which are known as current good manufacturing practices (CGMPs). Since the devices covered by the regulation are diverse, the document 21 CFR Part 820 does not provide specific instructions; instead, it describes a framework that should be used for quality assurance in design, development, production, installation, and servicing (Food and Drug Administration, “Quality System”). Under CGMPs, the manufacturer of QardioArm needed to establish individual requirements for the product to ensure its effectiveness and safety based on good judgment and provisions of the FDA.
Labeling regulations provide that all the products are properly labeled and supplied with necessary literature comprehendible to the intended user. For over-the-counter devices, FDA requires the package to include a clearly written name of the device, identification of quantity, and any warning statements applicable to the device (Food and Drug Administration, “21CFR801″). These requirements are easily comprehendible and provide instruction about how labeling should be done.
Medical device reporting regulation aims at the promotion of timely detection and corrections of problems by monitoring negative effects on medical devices (Food and Drug Administration, “21CFR803”). Under this regulation, the manufacturer needs to use the 3500A form to report that QardioArm caused severe injury or death (Food and Drug Administration, “21CFR803”). This form is also used to report any malfunctions that can potentially cause death or serious injury in users (Food and Drug Administration, “21CFR803”). Importers need to inform both FDA and manufacturers about cases of death and severe injury; however, in case of malfunctions, the importer needs to inform only the manufacturer (Food and Drug Administration, “21CFR803”). The medical personal also needs to report injuries and deaths both to the manufacturer and FDA, but no notification is required in case of malfunction.
In summary, QardioArm is a Class II device, which implies that is available over the counter. It was approved by the FDA through PMN in 2014 as it had analogs on the market. After the FDA’s approval, QardioArm needed to be registered in accordance with 21 CFR Part 807. Currently, the manufacturer of the device needs to comply with labeling, medical device reporting, and QS regulations. Moreover, the manufacturer needs to confirm the registration information annually.
Comparison of Two Regions
Comparative Chart
The regulations may differ in some aspects, depending on the region where a medical device is marketed. Table 1 below juxtaposes regulations applicable to QardioArm in the US and the EU. All the regulations for the US region are provided by FDA, while marketing of the device in the EU region is regulated by the European Medical Device Regulation (EU MDR) document created by the European Parliament and the Council on Medical Devices.
Table 1. Comparative analysis of regulation on QardioArm
Analysis
Table 1 above demonstrates that there significant differences in the regulation of QardioArm in the US and the EU. While the device belongs to essentially the same class in both regions and subject to no clinical trials, the device needs to go through different testing. In the US, the manufacturer of QardioArm needed to file PMN to the FDA to prove that the new product is similar to devices already on the market. According to EU MDR, QardioArm needed to go through Notified Body Conformity Assessment, which ensures that the device meets all the essential safety and effectiveness requirements (European Parliament). The process is much longer in the EU, as it incorporates more investigation measures. However, the process in the EU is associated with less criticism and increased control over possible adverse events and safety risks.
Another significant difference lies in post-approval modification reporting in the two regions. In the US, FDA requires to report changes only if they affect effectiveness and safety or if the device is redesigned to for new intended use (Food and Drug Administration, “21CFR807”). In Europe, the manufacturer needs to report a wider variety of changes under Annex VI of the EU MDR, including modification of software and changes in design (European Parliament). In other words, the process of reporting modifications is associated with more scrutiny in the EU, which provides fewer incentives for manufacturers to make slight changes in devices and market them as new ones.
Adverse event reporting requirements are also different in the two regions, as the EU Parliament puts more responsibility on the manufacturer than other stakeholders. In particular, FDA regulations oblige manufacturers, importers, and health care professionals to report cases of death or serious injury under 21 CFR 803 (Food and Drug Administration, “21 CFR 803”). At the same time, malfunctions that are not associated with health risks do not need to be reported (Food and Drug Administration, “21CFR803”). In Europe, all the responsibility is put on the manufacturer, as it is required to encourage other stakeholders to report any serious incidents not described in the side effects section (European Parliament). This implies that only the manufacturer needs to unexpected cases, and the range of such cases is increased in the EU.
Quality and manufacturing requirements and human factors and usability requirements are essentially similar in both regions. The only notable difference is that the enforcement of these requirements seems to be stricter in the EU, as the notified bodies are encouraged to conduct quality audits without prior notification (European Parliament). Therefore, in general, QardioArm is subject to more scrupulous regulations in the EU than in the US. On the one hand, it may be associated with increased quality of products and fewer devices on the market that have essentially similar functions. On the other hand, increased regulation may be associated with less possibility for innovation and decreased competition among medical device manufacturers.
Conclusion
Wearable diagnostic devices are becoming increasingly popular around the world, as they allow users to monitor their well-being and share gathered data with doctors. Wearable medical devices may transform medicine and healthcare in the nearest future as they become increasingly precise and popular. QardioArm is an excellent example of a wearable medical device that can help to manage hypertension and other heart-related conditions. The introduction of devices that help to address cardiovascular conditions is of extreme importance, as these diseases are the number one cause of death worldwide. An increased number of marketed medical devices requires strict regulation systems to ensure the effectiveness and safety of emerging technology. In the US, the FDA is the governing body that controls medical device marketing. While the FDA created a comprehensive regulatory pathway for medical devices, it is applicable only to the devices sold in the US. In Europe, the introduction and distribution of QardioArm are controlled by the EU MDR, which is significantly different from US laws. The analysis of regulations affecting QardioArm revealed that the policy concerning medical devices is stricter in the EU than in the US.
Works Cited
Chahine, Mirna et al. “Validation of BP Devices Qardioarm in the General Population and Omron M6 Comfort in Type II Diabetic Patients According to the European Society of Hypertension International Protocol (ESH-IP),” Medical Devices: Evidence And Research, Volume 11, 2018, pp. 11-20.
Centers for Disease Control and Prevention. “Facts about Hypertension.” CDC.
“Heart Disease Facts.” CDC.
European Parliament. “European Medical Device Regulation.” EUR-Lex.
Food and Drug Administration. “21 CFR 801.” FDA.
“21CFR803.” FDA.
“21CFR807.” FDA.
“510(kPre-marketet Notification.” FDA Database.
“Applying Human Factors and Usability Engineering to Medical Devices. FDA.
“Ki140067: Summary of Safety and Effectiveness.” FDA Database.
“Quality System (QS) Regulation/Medical Device Good Manufacturing Practices.” FDA.
Marr, Bernard. “The 9 Biggest Technology Trends that Will Transform Medicine and Healthcare in 2020.” Forbes, 2019. Web.
Pardo, Victoria et al. “The Qardioarm App in the Assessment of Blood Pressure and Heart Rate: Reliability and Validity Study.” JMIR Mhealth and Uhealth, vol. 5, no. 12, 2017, p. e198.
Phaneuf, Alicia. “Latest Trends in Medical Monitoring Devices and Wearable Health Technology.” Business Insider, 2020.
Qardio. “QardioArm.” GetQardio.
Qardio Support. “Taking a Measurement with QardioArm.” GetQardio.
“QardioArm User Manual.” ManualsLib.
Van Norman, Gail A. “Drugs, Devices, and the FDA: Part 2”. JACC: Basic to Translational Science, vol. 1, no. 4, 2016, pp. 277-287.
World Health Organization. “Cardiovascular Diseases.” WHO.
Zhang, Yuting et al. “What Matters the Adherence with BP 24-Hr Self-Monitoring Wearable Device among Hypertensive Patients? A Population-Based Survey”. Translational Behavioral Medicine, ibz069, 2019.