Introduction
The protein evolution rate and mechanisms are one of the crucial questions in evolutionary biology. It was estimated that this rate depends on the protein’s functional constraints. Modern methods of investigation, in particular, amino acid sequence, provide an opportunity to examine the dependence of protein functions on its primary structure (Zhang & Yang, 2015). It was early estimated that the rate of protein evolution significantly varies between different life domains, kingdoms, and even related species. Besides, different proteins of the same species also evolve with different speeds (Zuckerkandl & Pauling, 1965). Genetic investigations, performed in the last decades, have shown that the level of protein expression is important for the rate of its amino acid sequence evolution. It was shown that proteins with a high level of expression changes slowly (Park, Chen, Yang, & Zhang, 2013). Other determinants of protein evolution were also established, however, its functional importance was determined to be less significant (Zhang & Yang, 2015).
Traditionally, it was claimed that protein’s functions depend on not solely its sequence but also it’s tertiary (3D) structure (Schulz & Schirmer, 2013). This structure and, consequently, protein functions depend on the amino acid sequence of the polypeptide chain (Uversky, 2014). However, so-called intrinsically disordered proteins (IDPs) and proteins with intrinsically disordered regions (IDRs) were discovered and studied recently. Interestingly, this type of protein does not have a stable tertiary structure and exists in different conformations which made it more flexible and adaptive to various external conditions. This ability provides to these proteins certain advantages, in comparison to ordered proteins (Uversky, 2014).
Recent investigations have shown that IDPs are widely spread in nature among all the cellular and non-cellular living organisms. Besides, some of these proteins are associated with serious diseases such as cancer and neurological disorders (Dyson, 2016). Fully or partially disordered proteins accomplish such functions as cell metabolism regulation, cell-cell, and protein-protein interaction, molecular recognition, and gene expression regulation (Szalkowski & Anisimova, 2011). Thus, due to the importance of intrinsically disordered proteins, it is essential to study their unique structure, properties, functions, and evolution, using modern genetic and biochemical methods of investigation.
Intrinsically Disordered Proteins Properties
Traditionally, protein structural biology was based on the concept of the sequence-structure-function paradigm. According to this concept, protein functions are determined by its amino acid sequence. This paradigm was confirmed by numerous biochemical studies, provided on the wide range of proteins, including enzymes and receptors (Punta, Simon, & Dosztányi, 2015). However, novel researches, provided on IDPs and proteins with IDRs, changed this paradigm into the disorder-function paradigm. Figure 1 shows the difference between the two concepts and between ordered and disordered proteins. Miskei, Antal, and Fuxreiter (2016) postulated that disordered protein properties depend on their conformation which is determined by interaction with other proteins and external media characteristics.
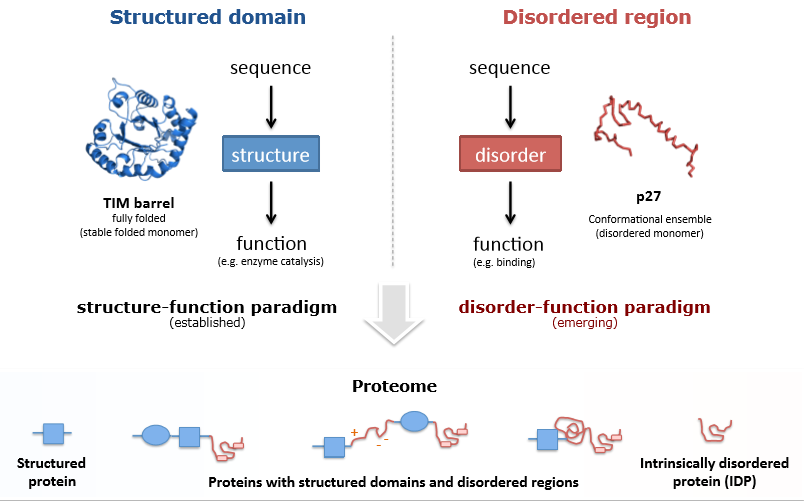
Disordered protein’s flexible structure determines its unique properties. It was established that these proteins are more stable in the environment and more resistant to its changing. Due to this ability, IDPs play the role of an essential hub in the network of molecular interactions (Wright & Dyson, 2015). The lack of a stable 3D structure determines the plasticity and high adaptive properties of IDPs. Due to these properties, disordered proteins are able to perform more complicated functions, in comparison to ordered proteins. In particular, IDPs play the leading role in the protein-protein interaction which is essential for numerous vital functions. (Cordeiroa et al., 2017).
IDPs properties are determined by the environment. This rule is common for all proteins because ordered protein properties also depend on environmental characteristics. Under some conditions (acidic or alkaline conditions, high temperature, and others), the process of protein denaturation occurs. However, IDPs are much more sensitive to environmental conditions. They react to even small environmental changes by adaptation of their conformation. Besides, the common property of IDPs is the ability to disorder-to-order transformation. Frequently, a single protein can take different structural conformations, depending on the structure of its protein-partner (Uversky, 2014).
The group of IDPs is not homogeneous. Some of the proteins are completely disordered, while others combine ordered and disordered regions (Figure 1). The length of disordered fragments varies among different proteins (Monastyrskyy et al., 2014). Disordered fragments can be associated with different protein regions: loop region, termini, and linking fragments. The shape of IDRs can also be different: coils, globules, and others. The level of the disorder can fluctuate from the completely unstructured to folded secondary structure with tertiary structure fragments (Punta et al., 2015).
It was estimated that IDPs amino acid sequence is unique and does not similar to the sequence of ordered proteins. In particular, IDPs and are characterized by the lower content of non-polar hydrophobic amino acids and the high content of polar net-charged residues (Das & Pappu, 2013). This specific characteristic of the sequence was also observed during the comparison of ordered and disordered regions of the same protein. For ordered regions, hydrophobic amino acids (phenylalanine, isoleucine, leucine, methionine, valine, tryptophan, and tyrosine) were more typical, while for disordered regions charged amino acids (aspartic acid, glutamic acid, lysine, glutamine, and serine) were preferred (Yu et al., 2016). Thus, it could be stated that intrinsically disordered proteins and proteins with intrinsically disordered regions have unique properties, different from ordered proteins.
Intrinsically Disordered Proteins Significance
It was established that IDPs and IDRs are widely spread among all living organisms. Modern investigations demonstrated that IDPs are common for eukaryotic (Szalkowski & Anisimova, 2011), archaeal and prokaryotic organisms (Yadav et al., 2016), and viruses (Chemes et al., 2012). Especially, IDPs are important for eukaryotic cells and multicellular organisms (Uversky, 2014). In these organisms, IDRs present at a higher rate: 36–42%, while prokaryotic cells contain just 7–13% of IDRs (Punta et al., 2015, p. 37).In general, approximately 10% of eukaryotic proteins are completely disordered, and approximately 50% of them have ODRs of different lengths (Szalkowski & Anisimova, 2011, p. 488).
IDPs play a major role in the processes of cell metabolism regulation, signaling, and molecules recognition. Some IDPs participate in the process of the RNA molecule’s rearrangement. Other disordered proteins act as splicing factors and assist in the processes of transcription and functional RNA formation. In viruses, in particular, in HIIV, IDPs participate in core formation, viral genes integration, expression, and replication. Besides, disordered proteins are responsible for recognition and interaction with different target molecules and new cell contamination (Peng et al., 2014). Thus, it could be postulated that IDPs and IDRs accomplish a wide range of significant biological functions.
Moreover, the malfunctioning of IDPs is associated with serious diseases, such as cancer and neurodegenerative disorders (Krzeminski et al., 2013). For example, CREB transcription factor is one of the proteins which are responsible for long-term memory formation. It was discovered that the malfunction of this protein is one of the reasons for Huntington’s disease. Besides, the wrong functioning of IDPs is associated with some types of cancer. For example, P53 tumor suppressor is an IDP which malfunction leads to carcinoma development. Normally, this protein suppresses tumor growth, and if it does not work properly, nothing prevents cancer (Soragni et al., 2016). Other disordered proteins are associated with neurodegenerative (in particular, Alzheimer’s and Parkinson’s diseases) and metabolic (in particular, type II diabetes), human diseases (Knowles et al. 2014).
Classification of Intrinsically Disordered Proteins
Different IDPs’ and IDRs’ classifications were developed, depending on their functions. In particular, Tompa (2012) divided all the IDPs’ and IDRs’ functions into several categories: molecular recognition, molecular aggregation, protein-protein interaction, modification of other molecules, and metabolic regulation. Another classification was developed by Gsponer and Babu (2012). According to the authors, IDRs’ functions could be classified into three groups: translation regulation and RNA modification, protein-protein interaction and binding, and molecular conformation changing and adaptation. Authors claimed that a single protein could contain several disordered regions and accomplish different functions. Van Der Lee et al. (2014) stated that six main classes of disordered protein regions could be determined: “entropic chains, display sites, chaperones, effectors, assemblers, and scavengers” (p. 6). All these classes of proteins accomplish different functions within the cell or organism. Figure 2 shows the functional classification of intrinsically disordered regions within the cell.
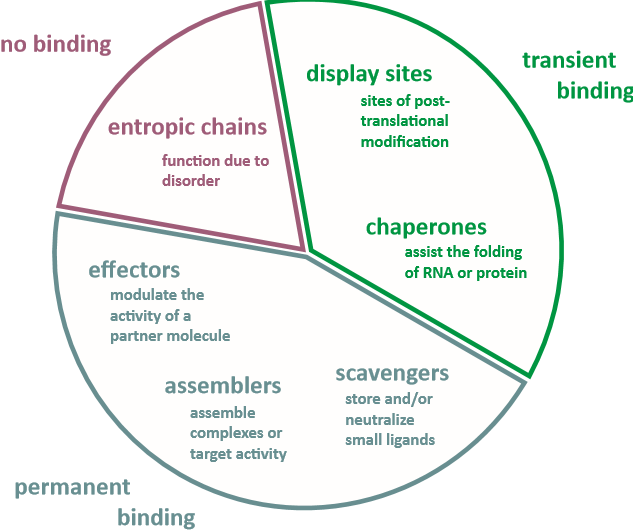
Entropic Chains
Entropic chains include linkers and spacers. These types of proteins are responsible for protein domains movement. They regulate distances between functional domains. The disordered structure is an essential characteristic of these regions’ functioning. As an example, 70 kDa subunit of replication protein A and microtubule-associated protein 2 could be cited. Entropic springs, such as the disordered region of titin protein, could be claimed as another example of entropic chains. This protein plays a significant role in over-stretching muscle recovery (Van Der Lee et al., 2014).
Display Sites
Post-translational modification (PTM) of proteins is important for its normal functioning. This process influences the protein transportation and localization within the cell, their stability, and the ability to accomplish certain functions (Beltrao et al., 2013). Display sites are the targets for PTM (Uversky, 2014). Comparing with ordered regions, IDRs have certain advantages as display sites because they are easily recognized, bound, and modified by PTM enzymes. These regions are a common substrate for several enzymes, such as kinases. It was experimentally shown that proteins display sites, prepared for phosphorylation, are enriched with IDRs (Wright, & Dyson, 2015). As another example of proteins with disordered display sites, histones could be cited (Van Der Lee et al., 2014).
Chaperones
Chaperones were determined as proteins that allow DNA, RNA molecules, and proteins to reach their stable functional states. It was shown that approximately 50% of nucleic acid chaperones and nearly a third part of protein chaperones contain disordered regions. This tendency could be explained by the flexibility of IDRs which is suitable for chaperones functioning. Firstly, IDRs are able to interact with a wide range of other proteins. Thus, one single chaperon is capable to assist different protein modifications. The second advantage is the speed of reaction. It is suitable in case of emergency, for example, for preventing the toxic protein aggregation formation. Moreover, thermodynamic characteristics of IDRs are suitable for repetitive processes of binding-unbinding with the target substrate. The IDRs action allows lowering the demand for ATP for these processes. However, the exact molecular mechanisms of these processes still remain unclear. As an example of IDR-reach chaperone, redox-regulated chaperone Hsp33 could be cited (Van Der Lee et al., 2014).
Another functional group of IDRs in this category is RNA- and DNA- binding proteins. These proteins present in the nucleus and participate in the process of gene transcription regulation. It was shown that approximately half of the transcription factors are proteins with disordered regions (Peng et al., 2014). As an example, RNA-binding La protein with disordered C-terminal domain could be cited. This domain allows this single protein interaction with different non-coding RNA precursors, protecting the molecule from cellular nucleases.
Effectors
Effectors are proteins that modify the activity of other proteins (Pang & Zhou, 2016). After the target binding, the disordered region of effectors transforms into order. This process is called “coupled folding and binding” (Van Der Lee et al., 2014, p. 7). P21 and P27 proteins could be cited as examples of such effectors. These mammals proteins regulate some of the cyclin-dependent kinases (Cdk) in cells. These kinases are responsible for cell biochemical cycles. P21 and P27 enhance the activity of Cdk4, and decrease the activity of Cdk2 enzymes. Calpastatin, which binds and inhibits calpain, is another example of effectors. After binding the target protein, the disordered region of calpastatin reversible transforms into the ordered globular fragment (Rao et al., 2014). The ability of these effectors to interact with proteins is determined by its flexibility due to disordered regions’ presence.
Assemblers
Assemblers could be defined as proteins that are responsible for molecules binding and formation of protein complexes (Wallace, Budnik, & Drummond, 2015). The percentage of IDPs or proteins with IDRs among all assemblers is high. In particular, IDPs play an important role in the ribosome organization, immune cell activation, and transcription initiation. To accomplish all these functions, it is essential to combine several different proteins into a complex. Disordered regions are capable to perform it more effectively than ordered. Due to its unique flexibility, a single IDR is able to bind several different target proteins. Thus, the intrinsic disorder is the commonly spread feature for assemblers which play the role of protein network hubs (Van Der Lee et al., 2014). As the example, the axis inhibition scaffold protein could be cited. This protein binds together β-catenin, casein kinase Iα, and glycogen synthetase kinase 3β and forms an effective functional complex which is essential for β-catenin destruction.
Scavengers
Scavengers are special proteins which store and neutralize small active molecules (Kainz, & Reiser, 2014). These proteins frequently are enriched with IDRs which significantly increase their effectiveness. Chromogranin A and casein are the examples of such proteins. Chromogranin’s main function is to store ATP and adrenaline molecules in inactive forms in the adrenal gland medulla. Casein is able to bind and store calcium phosphate (Van Der Lee et al., 2014).
Altogether, the unique disordered protein’s structure determines several advantages, comparing with ordered polypeptides. Among them, high adaptive properties, compact functional regions, their high density within the protein molecule, and specific binding to the variety of target molecules could be claimed. These properties are the reasons of IDPs leading roles in such essential processes as gene transcription and expression regulation, signaling processes, molecular interaction, and others (Punta et al., 2015). Due to the wide range of disordered proteins functions, their investigation is a highly-important and fast-developing field of research. Novel experimental data about IDPs makes scientific knowledge about molecular mechanisms of protein functioning and interaction deeper. In recent time, the crucial sequence-structure-function paradigm of protein structural biology was reconsidered due to discovering of unique characteristics of IDPs. Disordered protein studying is a complicated field of research which requires both genetic and biochemical modern methods of investigation applying (Uversky, 2014).
Intrinsically Disordered Proteins Evolution
Relatively new methods of the determination of the proteins amino acid sequence provided an opportunity to investigate homologous proteins in different species and to estimate the rate of proteins evolution (Zhang & Yang, 2015). These researches are crucial for evolution mechanism understanding and reconstruction. It was early estimated that rates of different proteins evolution could be vastly different even for the same species (Zuckerkandl & Pauling, 1965). This difference was explained by the neutral theory of protein evolution. According to this theory, the rate of sequence evolution depends on the rate of neutral mutation. Positive mutations were not taken into account because they were considered to be too rare to affect the evolution rate. The neutral theory of protein evolution postulated that the rate of evolution depends on functional constraints of the protein: the stronger these constraints are, the lower the rate of sequence changing. However, no strict definition of the functional constraint exist, which was considered to be the limitation of this theory (Van Der Lee et al., 2014).
After discovering IDPs and IDRs, the question about their evolution rate appeared. According to Brown et al. (2010), Markov amino acid substitution model could be used for estimation of the rates of ordered and disordered proteins evolution. This model allows comparing frequencies of amino acids changes and estimation the average rate of protein evolution. The authors determined that the average rate of disordered proteins and regions evolution is higher than the rate of ordered fragments sequence changing. These results were confirmed by the investigation of Dyson (2016). The author studied the scoring matrix of proteins with IDRs and concluded that disordered regions are evolving faster than ordered within the same protein molecule.
Thus, it could be stated that disordered regions generally evolve faster. These regions are characterized by the higher rate of amino acid substitutions, insertions, and deletions, in comparison to ordered regions. This tendency could be explained by the lack of constraints, based on the connection between sequence and structure. It was claimed that these constraints limited the evolution of ordered protein. For ordered regions, a single amino acid substitution might cause the structural instability and functional disability. Thus, in this case, this changing might be excluded by the purifying selection. The situation is different with disordered regions. For them, the protein structure does not directly depend on amino acid sequence. Thus, this sequence could be changed without serious consequences for protein functioning (Van Der Lee et al., 2014).
A recent investigation, provided by Szalkowski & Anisimova (2011), established that for more than a half of IDPs, disordered regions are less conserved than the rest of the protein chain. However, it was shown that this general tendency has many exceptions. For approximately 15% of studied IDPs, the rates of ordered and disordered fragments evolution were not significantly different. For example, it was estimated that ordered and disordered regions of associated with cancer E7 oncoprotein of papillomavirus evolve with the similar rate (Chemes et al., 2012), while disordered domains of calcineurins, topoisomerase, S4 ribosomal protein and many others polypeptides evolve faster (Uversky, 2014). Furthermore, for approximately 27% of proteins, the rate of disorder regions evolution was significantly lower than the rate of ordered regions evolution (Szalkowski & Anisimova, 2011). These studies highlighted that protein evolution remains unclear and requires further investigations with the purpose to estimate its mechanisms.
It was established that the amino acid sequence conservation of disordered regions depends on their functions. Different functional classes of IDRs evolve with the different speed (Uversky, 2014). Some IDRs, for example, human α-synuclein and flagellin, were determined to be highly conserved. Generally, more functionally important fragments are more conserved. IDPs with the low rate of changing are common for different species and even domains of life. Moreover, within the same IDR the speed of evolution could be different: some amino acid sequences evolve faster while others seem to be more conserved. All these data highlighted the high dependence between evolution rate and functions of the whole molecule or its parts.
It was also shown that the rate of protein evolution depends on the complexity of organisms. Generally, more complex organisms have a higher percentage of IDPs and IDRs among other polypeptides, due to more complex functions presence. It was predicted that 33% of eukaryotic proteins contain IDR, in comparison with archaeal (2%) and bacterial (4%) cells (Ward et al., 2004). The content of disordered regions varies among different prokaryotic and eukaryotic species. Furthermore, it was estimated that the difference of IDR among human’s chromosomes distribution exists (Van Der Lee et al., 2014). Viruses’ studies have shown that viruses with the small RNA genome contained the higher rate of IDRs than prokaryotic cells. For viruses, it was supposed that the capacity of a single disordered protein to interact with different target molecules is essential for reducing the genome size of viral particles (Chemes et al., 2012).
Interestingly, for different life kingdoms, the variation of IDRs preferable functions exists. For eukaryotic cells and viruses, IDRs are mainly responsible for short-time protein-protein interaction for the signaling and regulation processes. In contradiction to, bacterial and archaeal cells use proteins with disordered regions for the formation of long-lasting complexes of molecules. Thus, it could be concluded that not only amino acid sequence but also functions of IDPs and IDRs evolved differently among different life domains and kingdoms (Van Der Lee et al., 2014).
It was stated that IDPs provide the functional benefits for more complex organisms, in particular, multicellular eukaryotic species. Although IDPs and IDRs are present in prokaryotic cells, their percentage increased significantly in eukaryotic organisms. Thus, it could be concluded that disordered regions are the rather novel evolutionary invention, and the further increase of its presence within cells could be expected in future (Romero et al., 2006). In contradiction to this hypothesis, it was supposed that pre-cell polypeptides, which appeared in the primordial soup on the ancient Earth, were disordered. Furthermore, it was observed that the rate of Darwinian selection was higher for disordered regions, comparing with the selection of structured conformations. It is possible that certain percentage of IDPs is required for the increase of genetic variation and adaptive properties of the cell or organism (Uversky, 2014). Moreover, the author suggested that the ability of disordered proteins to perform complex functions led to the development of more complex organisms. It was a basis for the eukaryotic cells and, later, multicellular organisms appearance.
Modern Methods of Disordered Proteins Investigation
Nowadays, modern experimental and theoretical approaches for IDPs and IDRs studying is developing due to the existing needs for the analysis of proteins sequence, structure, properties, molecular mechanisms of functioning and interaction with other molecules (Uversky, 2014). In general, two types of investigation methods are applied: biochemical and genetics. Biochemical methods(x-ray crystallography, nuclear magnetic resonance, small-angle scattering, and others) allow studying of protein’s properties in vitro and in vivo, while genetic methods provide the information about protein-coding genes nucleotide and protein amino acid sequences. All these methods have their strengths and limitations. Thus, to obtain clear data, different approaches should be used simultaneously.
Nuclear Magnetic Resonance (NMR) is a method for protein structural conformation investigation. This method is the most commonly used technique for ordered proteins studying. However, it has certain limitations for the investigation of IDPs and IDRs. In particular, it is difficult to estimate proper sizes and shapes of proteins. To complement the NMR, various other methods are used, including Small-Angle Scattering (SAS) of X-rays (SAXS) or Neutrons (SANS). SAS technique is considered to be low-resolute but sensitive to conformation fluctuations. However, due to the significant structural variability of IDPs, transformation of obtained average experimental data into the proper information about protein 3D structure is rather difficult and requires the special computational approaches development (Cordeiro et al., 2017).
One of the widespread biochemical methods of investigation is X-ray crystallography. It is a method which allows determining the atomic and molecular structure of ordered crystals, using the diffraction of x-rays (Barends et al., 2014). In protein structure, obtained by this method, disordered regions are usually framed by ordered fragments. However, the IDRs’ structure could not be exactly defined. It was supposed that conformational flexibility makes their tertiary structure undefined by x-ray crystallography. Thus, if such results are obtained during the protein in vitro investigation, it could be assumed that this protein has disordered regions. Another sign of IDRs, which could be identified by x-ray crystallography, is different distances between residues, found in different protein chains. However, it should be mentioned that disordered structures could temporary be transformed into ordered, as a result of interaction with their target molecules (RNA, DNA, other proteins). Therefore, under these conditions, x-ray studying might mistakenly identify ordered structures instead of disordered regions (Punta et al., 2015).
All proteins were evolved inside the cells and were adopted to act in the specific conditions which are different from the external environment. This difference provides certain difficulties for the IDPs’ investigations. All the biochemical in vitro experiments should be performed under specific conditions with certain macro- and microelements concentrations in the solution, the certain temperature, and other characteristics (Theillet et al., 2014). These conditions are close to those which are observed in cells. However, the intercellular environment is changeable and depends on the particular needs of the cell. Correspondingly, IDPs properties could also be changed in response to the environment characteristics. Thus, biochemical methods of investigations could provide only limited data according to the IDPs properties and functions (Uversky, 2014).
Recent investigations have shown that IDPs and IDRs have the specific amino acid sequence. In particular, they are characterized by low content of non-polar amino acids and low complexity as well as by the high content of net-charged residues. Thus, it could be supposed that disordered regions might be predicted by reading protein amino acid sequence. To predict this region presence, it is possible to develop special computational approaches. In current time, such approaches are widely used in modern investigations of disordered proteins. To validate the information, obtained by computational models application, experimental data should be used (Peng et al., 2013).
Commonly, to determine exact 3D models of disordered proteins, data about residue-specific conformational structures, obtained from crystallographic structures databases, is used. To create such model, researches compare existing data about the sequence-conformation dependence with the amino acid sequence of particular IDP. Generally, amino acid sequence determines the secondary conformation of the protein and the interaction of separated domains. This interaction is responsible for 3D conformation. Besides, it is important to develop the energy model which might take into account the force-field interaction within the chain. As the examples of such models, Molecular Dynamics or Monte-Carlo simulations could be cited. These methods are appropriate to determine the conformational structure of disordered proteins. However, the lack of the protein sequence information is the main limitation for this method usage. It is essential to accumulate data according to the sequence-conformations dependence (Punta et al., 2015).
The lack of the experimental data highlights the necessity of bioinformatics approaches development for IDPs and IDRs prediction. These approaches are important for IDPs structure, properties, and significance understanding. During the last decades, the information about different IDPs sequences, structure, and functions was accumulated, and the necessity of organized databases appeared. The first database was The Database of Protein Disorder (DisProt) (Vucetic et al., 2005). It contains the information about disordered protein fragments of from 30 to over 18,000 amino acid residues. However, the size of this database is limited. Intrinsically Disordered Proteins with Extensive Annotations and Literature (IDEAL) is another available IDPs database which contains the information about sequence and structure variations of proteins (Fukuchi et al., 2015). Recently, two more IDPs databases appeared: MobiDB (Di Domenico et al., 2012) and D2P2 (Oates et al., 2013). These databases combine the available from other database protein sequence information to predict the IDRs presence within the protein chain.
Conclusion
Traditionally, protein structural biology was based on the sequence-structure-function paradigm which postulated the dependence of protein functions on the sequence-determined 3D structure. However, the investigations, performed during recent decades, discovered the alternative type of proteins: completely intrinsically disordered proteins or proteins with intrinsically disordered regions. IDPs have unique properties, which determine their functioning. The main characteristic of these proteins is the lack of the 3D structure. It means that the conformation of the particular protein molecule depends on environmental conditions and the process of binding with target molecules (DNA, RNA, or other proteins). According to their properties, IDPs accomplish the wide ranges of functions. They are used in such process as molecules recognition, signaling, gene expression regulation, and others. Due to their functions, six categories of proteins with disordered regions could be distinguished: entropic chains, display sites, chaperones, effectors, assemblers, and scavengers.
It was estimated that these proteins are widespread among all the life domains and kingdoms. However, they are especially typical for eukaryotic organisms, in particular, for humans. Moreover, these proteins are associated with some serious diseases in humans, such as cancer and neurodegenerative diseases. These diseases appeared if certain proteins in human body started functioning in the wrong way. The rate of evolution of ordered and disordered proteins is also different. Generally, IDPs and proteins with IDRs evolve faster than ordered fragments. It was also supposed that primary polypeptides were disordered and that these proteins allowed the evolution of more complex living organisms.
Thus, due to the high importance of disordered proteins, different methods of investigation were developed. Traditional methods of structural biology, such as nuclear magnetic resonance, small-angle scattering, and X-ray crystallography, have certain limitations in disordered region studying due to the lack of 3D structure. Therefore, computational methods and bioinformatics approaches were developed to predict disordered regions in proteins and their properties. These methods are based on protein amino acid sequences. Bioinformatics databases with the information about sequences and properties of IDPs and IDRs are available nowadays. Thus, it could be concluded that disordered proteins and regions studying is the fast-developing and highly important field of investigation.
References
Barends, T. R., Foucar, L., Botha, S., Doak, R. B., Shoeman, R. L., Nass, K.,… & Schlichting, I. (2014). De novo protein crystal structure determination from X-ray free-electron laser data. Nature, 505(7482), 244-247.
Beltrao, P., Bork, P., Krogan, N. J., & van Noort, V. (2013). Evolution and functional cross‐talk of protein post‐translational modifications. Molecular Systems Biology, 9(1), 714-726.
Brown, C. J., Johnson, A. K., & Daughdrill, G. W. (2010). Comparing models of evolution for ordered and disordered proteins. Molecular Biology and Evolution, 27(3), 609-621.
Chemes, L. B., Glavina, J., Alonso, L. G., Marino-Buslje, C., de Prat-Gay, G., & Sánchez, I. E. (2012). Sequence evolution of the intrinsically disordered and globular domains of a model viral oncoprotein. PLoS One, 7(10), 47661-47669.
Cordeiro, T. N., Herranz-Trillo, F., Urbanek, A., Estaña, A., Cortés, J., Sibille, N., & Bernadó, P. (2017). Small-angle scattering studies of intrinsically disordered proteins and their complexes. Current Opinion in Structural Biology, 42, 15-23.
Das, R. K., & Pappu, R. V. (2013). Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proceedings of the National Academy of Sciences, 110(33), 13392-13397.
Di Domenico, T., Walsh, I., Martin, A. J., & Tosatto, S. C. (2012). MobiDB: A comprehensive database of intrinsic protein disorder annotations. Bioinformatics, 28(15), 2080-2081.
Dyson, H. J. (2016). Making sense of intrinsically disordered proteins. Biophysical Journal, 110(5), 1013-1016.
Fukuchi, S., Amemiya, T., Sakamoto, S., Nobe, Y., Hosoda, K., Kado, Y.,… & Ota, M. (2014). IDEAL in 2014 illustrates interaction networks composed of intrinsically disordered proteins and their binding partners. Nucleic Acids Research, 42(1), 320-325.
Gsponer, J., & Babu, M. M. (2012). Cellular strategies for regulating functional and nonfunctional protein aggregation. Cell Reports, 2(5), 1425-1437.
Kainz, Q. M., & Reiser, O. (2014). Polymer-and dendrimer-coated magnetic nanoparticles as versatile supports for catalysts, scavengers, and reagents. Accounts of Chemical Research, 47(2), 667-677.
Knowles, T. P., Vendruscolo, M., & Dobson, C. M. (2014). The amyloid state and its association with protein misfolding diseases. Nature Reviews Molecular Cell Biology, 15(6), 384-396.
Krzeminski, M., Marsh, J. A., Neale, C., Choy, W. Y., & Forman-Kay, J. D. (2013). Characterization of disordered proteins with ENSEMBLE. Bioinformatics, 29(3), 398-399.
Miskei, M., Antal, C., & Fuxreiter, M. (2016). FuzDB: Database of fuzzy complexes, a tool to develop stochastic structure-function relationships for protein complexes and higher-order assemblies. Nucleic Acids Research, 45(1), 228-235.
Monastyrskyy, B., Kryshtafovych, A., Moult, J., Tramontano, A., & Fidelis, K. (2014). Assessment of protein disorder region predictions in CASP10. Proteins: Structure, Function, and Bioinformatics, 82(2), 127-137.
Oates, M. E., Romero, P., Ishida, T., Ghalwash, M., Mizianty, M. J., Xue, B.,… & Dunker, A. K. (2013). D2P2: Database of disordered protein predictions. Nucleic Acids Research, 41(1), 508-516. Web.
Pang, X., & Zhou, H. X. (2016). Mechanism and rate constants of the Cdc42 GTPase binding with intrinsically disordered effectors. Proteins: Structure, Function, and Bioinformatics, 84(5), 674-685.
Park, C., Chen, X., Yang, J. R., & Zhang, J. (2013). Differential requirements for mRNA folding partially explain why highly expressed proteins evolve slowly. Proceedings of the National Academy of Sciences, 110(8), 678-686.
Peng, Z., Oldfield, C. J., Xue, B., Mizianty, M. J., Dunker, A. K., Kurgan, L., & Uversky, V. N. (2014). A creature with a hundred waggly tails: Intrinsically disordered proteins in the ribosome. Cellular and Molecular Life Sciences, 71(8), 1477-1504.
Punta, M., Simon, I., & Dosztányi, Z. (2015). Prediction and analysis of intrinsically disordered proteins. Structural Proteomics: High-Throughput Methods, 1261, 35-59.
Rao, M. V., McBrayer, M. K., Campbell, J., Kumar, A., Hashim, A., Sershen, H.,… & Nixon, R. A. (2014). Specific calpain inhibition by calpastatin prevents tauopathy and neurodegeneration and restores normal lifespan in tau P301L mice. Journal of Neuroscience, 34(28), 9222-9234.
Romero, P. R., Zaidi, S., Fang, Y. Y., Uversky, V. N., Radivojac, P., Oldfield, C. J.,… & Dunker, A. K. (2006). Alternative splicing in concert with protein intrinsic disorder enables increased functional diversity in multicellular organisms. Proceedings of the National Academy of Sciences, 103(22), 8390-8395.
Schulz, G. E., & Schirmer, R. H. (2013). Principles of protein structure. New York, NY: Springer.
Soragni, A., Janzen, D. M., Johnson, L. M., Lindgren, A. G., Nguyen, A. T. Q., Tiourin, E.,… & Pellegrini, M. (2016). A designed inhibitor of p53 aggregation rescues p53 tumor suppression in ovarian carcinomas. Cancer Cell, 29(1), 90-103.
Szalkowski, A. M., & Anisimova, M. (2011). Markov models of amino acid substitution to study proteins with intrinsically disordered regions. PloS One, 6(5), 488-496.
Theillet, F. X., Binolfi, A., Frembgen-Kesner, T., Hingorani, K., Sarkar, M., Kyne, C.,… & Elcock, A. H. (2014). Physicochemical properties of cells and their effects on intrinsically disordered proteins (IDPs). Chemical Reviews, 114(13), 6661-6714.
Tompa, P. (2012). Intrinsically disordered proteins: A 10-year recap. Trends in Biochemical Sciences, 37(12), 509-516.
Uversky, V. N. (2014). Intrinsically disordered proteins. New York, NY: Springer.
Van Der Lee, R., Buljan, M., Lang, B., Weatheritt, R. J., Daughdrill, G. W., Dunker, A. K.,… & Kim, P. M. (2014). Classification of intrinsically disordered regions and proteins. Chemical Reviews, 114(13), 6589-6631.
Vucetic, S., Obradovic, Z., Vacic, V., Radivojac, P., Peng, K., Iakoucheva, L. M.,… & Newton, C. D. (2005). DisProt: A database of protein disorder. Bioinformatics, 21(1), 137-140.
Wallace, E., Budnik, B., & Drummond, D. A. (2015). Widespread protein assembly as a stress response. The FASEB Journal, 29(1), 563-511.
Ward, J. J., Sodhi, J. S., McGuffin, L. J., Buxton, B. F., & Jones, D. T. (2004). Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. Journal of Molecular Biology, 337(3), 635-645.
Wright, P. E., & Dyson, H. J. (2015). Intrinsically disordered proteins in cellular signalling and regulation. Nature Reviews Molecular Cell Biology, 16(1), 18-29.
Yadav, B. S., Singh, S., Kumar, P., Mathur, D., Meena, R. K., Agrawal, R. K., & Mani, A. (2016). Bioinformatics-based study on prokaryotic, archaeal and eukaryotic nucleic acid-binding proteins for identification of low-complexity and intrinsically disordered regions. Frontiers in Life Science, 9(1), 2-16.
Yu, J. F., Wu, E. S., Wang, C. L., Wang, H. M., & Wang, J. H. (2016). Classification of ordered/disordered regions of intrinsically disordered proteins based on comprehensive sequence analysis and Chou’s pseudo amino acid composition method. MATCH Commun Math Computer Chem, 75, 417-430.
Zhang, J., & Yang, J. R. (2015). Determinants of the rate of protein sequence evolution. Nature Reviews Genetics, 16(7), 409-420.
Zuckerkandl, E., & Pauling, L. (1965). Molecules as documents of evolutionary history. Journal of Theoretical Biology, 8(2), 357-366.