Introduction
This particular set of experiment was aimed at hybridizing nucleic acid and then characterizing the hybridized nucleic acid, which is a very important process in gene detection. This analysis helps us gain more insight into the structures, arrangement, and genes expression. Nuclei acid hybridization is a molecular technique that is widely used in analysis of complicated long chain nucleic acid. The technique facilitates easy and accurate recognition or classification of sequences/ series that are related to explicit probe series. A specific or particular sequence of the nuclei acid can be recognized from a heterogeneous/varied array if deoxyribonucleic acid portions are first size cut into small sizes using technique of agarose gel electrophoresis. Then the fractioned DNA sequences are then moved to deoxyribonucleic acid binding membrane in a lone stranded shape (Kinjo & Rigger, 1995).
The probe sequence /series is then labeled using radioactive tagging technique, and then modeled into a single strand. The resulting tagged probe sequence is then transferred into a solution and given time wash over the casing/membrane that has the target and other series. Under the suitable conditions, the probe will only anneal to those series/sequences on the membrane that are similar in terms of structure, same among others to it (Eigen & Rigler, 1994).
In this research, a plasmid sub-clone approach is used to classify a related series or sequences from a constraint digest/assimilate of a huge recombinant plasmid. The deoxyribonucleic acid (DNA) that was used in this particular experiment as a probe is a control portion from the plasmid pMAQ105. This restriction portion is then recovered by applying gel purification technique (Rigger, 1995). The DNA used is this exercise for probing is an amalgamation of plasmids pMAQ28 and R388. The amalgamated plasmids measures 37 kb in total (Zeiss, 2010). The appropriate restriction map that is applicable to this experiment is as shown below:
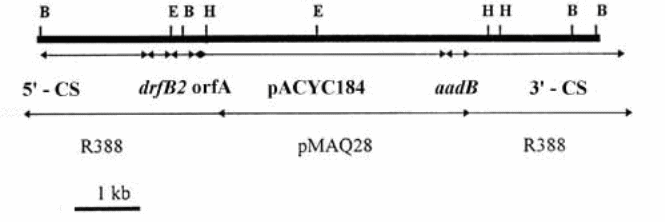
Key: Relevant restriction sites: B = BamH1, E = EcoR1, H = HindIII
Apparatus and Procedure
Reagents
TE = 10 mM Tris pH 8.0, 1 mM EDTA
Depurination solution: 0.25 M HCl
20X SSC = 0.3 M Sodium citrate, 3 M NaCl, pH7.0
Pre/Hybridization buffer: 2X SSC, 0.5% (w/v) blocking agent, 5% (w/v) Dextran sulphate, 0.1% (w/v) SDS
Buffer 1: 0.10 M Tris-HCl, pH 7.5, 0.15 M NaCl
Buffer 2: 0.10 M Tris-HCl, pH 7.5, 0.15 M NaCl, 0.5% (w/v) Blocking Reagent
Buffer 3: 0.10 M Tris-HCl pH 9.5, 0.10 M NaCl
Antibody Conjugate Solution: 1/1000 (v/v) Antifluorescein-AP conjugate in Buffer 2
Digest 1 B = BamH1, E = EcoR1, H = HindIII was set up for restriction digests of amalgamated DNA – pMAQ28/R388. The test tubes were then labeled, kept warm at 37 oC for two hours before being removed and placed in order in a rack. From the sheep provided, the position of each tube on the rack was entered where the three samples were placed. The technician then loaded the specimen, ran them on agarose gel, and then put back on the rack in the order in which they were run
Step 2
The gel was photographed followed by identifying the tracks onto which our specimen had been loaded. After identification, a small wedge of gel was trimmed off at the top right corner. The obtained wedge gel was then placed in a tray and 0.25m hydrochloric acid (depurination solution). The solution was left top to stand for 20 minutes under moderate stirring. After 20 minutes, the solution was tipped off and then washed with distilled water and the gel put in 0.4M sodium hydroxide (NaOH). As the gel was soaking in basing solution, zeta-probe and Whatman paper were cut into three pieces to the same size as the size of the gel. A lot of paper towel was cut to the same size as the size of the gel soaked and then piled together to a height of 6 cm high. The filter was then wetted with 0.4 M sodium hydroxide and the experiment apparatus for capillary transfer set as shown below to be transferred in 0.4M sodium hydroxide.
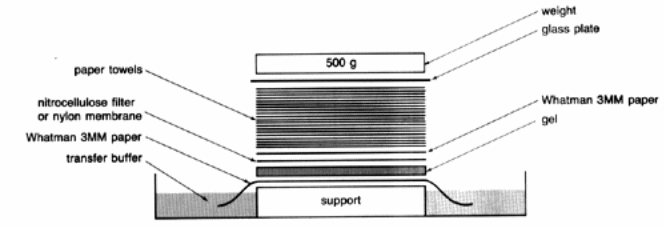
After completion of transfer of O/N at room temperature, the membrane was then enfolded in cling wrap and then kept in a freezer.
Probe Preparation
The probe sequence used in this experiment was derived from plasmid pMAQ105. Concentrated(undiluted) pMAQ105 specimen prepared earlier was used in this part. EcoR1 digest of pMAQ105 was set up and kept warm at 37oC for two hours. The digested specimen was then put on the rack and the precise position recorded on sheet in reading readiness for loading on a gel in the coming experiment.
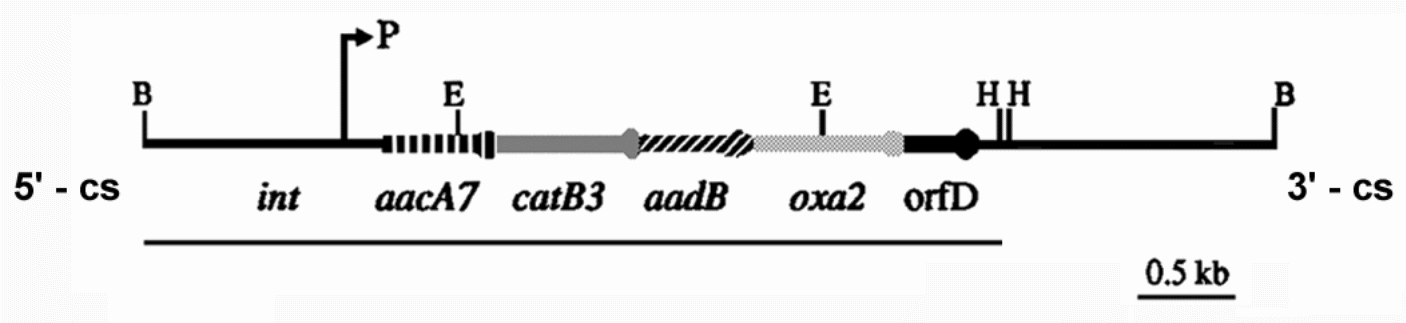
pMAQ105 insert
Relevant restriction sites: B = BamH1, E = EcoR1, H = HindIII
Membrane Preparation
A filter was recovered and then soaked for 30 seconds in 2XSSC, followed by inserting the membrane prepared in a hybridization bag, 15ml of pre-hybridization buffer solution and 750 μg DNA carrier was added and the n the bag carefully sealed as we ensured that all the air in the bad was purged. The bag was then incubated for at least 2 hours at 60ºC. Laboratory technician both prepared the probe of which we used in the subsequent experiments and placed the gel on the UV light box. The track on which our specimen was placed and the band that stands for the EcoR1 portion that was to be used as the probe was identified. Using the provided razor blade, portions of the gel under consideration was cut into small pieces and then put in an Eppendorf tube. The weight of the gel put in the tube was determined by weighing an empty tube followed by weighing tube with gel and then computing the difference.
10 μl of membrane binding solution was added to every 10mg of gel cut above, then vortexed before being incubated at 60ºC for roughly 10 minutes to ensure complete dissolution of the gel slices. A mini column was then inserted into collection tube followed by transferring the dissolved gel mixture in the assembled mini column. The solution was kept at room temperature for about 1 minute followed by centrifuging the solution at the highest speed of the centrifuge machine for one minute. The flow was disposed off and then the mini column reinserted into the collection tube. 700 μl of membrane wash solution was added, centrifuged at the greatest speed for 5 minutes, and then discarded. Again, 500 μl of membrane wash solution was added and then the assembly centrifuged for 5 minutes at the top speed. The contents of the collection tube were discarded and then column assembly centrifuged again for another one minute with micro centrifuge lid not tightened to give room for evaporation of ethanol that may have remained in the column. The mini column was the transferred into a clean 1.5ml Eppendorf tube followed by adding 30 μl water free of nuclease and then given about two minutes before centrifuging the solution for 1 minute at top speed. The contents were then discarded and the tube clearly labeled.
DNA specimen was boiled for 10 minutes band then instantly put on ice for 5 minutes. The solution was then spanned briefly; a labeling mixture that contained random Primers and reaction buffer mix, fluorescein nucleotide mix, and Klenow fragment was added to DNA solution followed by incubating the solution for one hour at 37ºC. the reaction was halted by boiling the mixture for 10 minutes at 60ºC.
To mix the membrane and the probe, 300 μl of hybridization buffer solution and 250-μg DNA carrier was carefully added to the probe. The probe was denatured by heating the solution for 5 minutes and then chilling it on ice. Denatured tagged probe was added to equilibrated 15 ml of hybridization buffer at 60ºC. pre-hybridization buffer solution contained in the bag was emptied and then probe DNA mix placed in this bag. The solution was then Hybridized at 60ºC.
Membrane was removed from the bag and then put in a g=big weigh boat tray. The membrane on the tray was the covered with 2 X SSC, 1% SDS and kept warm at 60ºC for 15 minutes. The liquid was tipped off and the same procedure repeated using 0.2X SSC, 0.1% SDS. After this, the liquid was again tipped off and the membrane covered in buffer solution at 15ºC for 5minutes under constant stirring. Membrane pores were blocked by covering the membrane in a buffer solution 2 for one hour. The membrane was then put in antibody conjugate solution and kept warm for one hour under moderate agitation. The membrane was washed 3 times for 5 minutes each in buffer 1. Followed by rinsing again the membrane two times for 5 minutes each in buffer 3.
Chemiluminescence Step
The membrane was transferred to a clean weigh boat tray, then covered with CDP star and the entire set up kept warm for 5 minutes at 37oC. Any excess solution was removed by wiping using tissue paper. This was wrapped in plastic wrapper by carefully folding the all plastic wrap on the “wrong” side of the membrane. The membrane was exposed to the film followed by developing the film.
Autograph was examined. The photo of southern gel was examined to determine which area of pMAQ28/R388 and pMAQ105 was homologous
References
Eigen, M. & Rigler, R. (1994). Sorting single molecules: Applications to diagnostics and evolutionary biotechnology. Proc. Natl. Acad. Sci., 91, 5740-5747.
Kinjo, M. & Rigger, R. (1995). Ultrasensitive hybridization analysis using fluorescence correlation spectroscopy. Nuc. Acids. Res. Journal, 10, 1-14.
Marmur, J. & Doty, P. (1962). Basic (bas) calculations were carried as described. Journal of Molecular Biology, 5, 109-118.
Rigger, R. (1995). Fluorescence correlations, single molecule detection, and large number screening. Applications in biotechnology. Journal of Biotechnology, 41, 177-186.
Sigma-Aldrich. (2011). Oligos melting temperature.
Zeiss, C. (2010). Analysis of nucleic acid hybridization and determination of kinetic parameters. Web.